Date: Fri, 12 Jun 2020 18:08:54 +0000
Hello All,
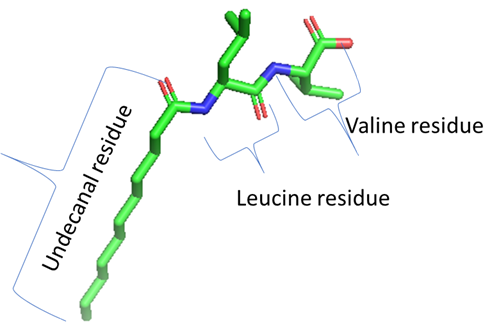
I am following up on a previous email that was sent by my research advisor (Dr. Eugene Billiot). I have a few questions regarding the application and creation of mixed force fields within a molecule. As mentioned previously in this thread, we are trying to create a micelle using undecyl-leucine-valine (ULV). Previous computational studies on ULV have entirely gaff force fields applied. However, the results produced are not well-suited for our particular study thus we would like to apply mixed force fields to our system. We would like to apply the gaff force field to the hydrocarbon chain (undecanal residue), and the protein.ff14SB force field to the dipeptide (leucine-valine) head group of the surfactant. The structure is displayed below (also attached to this email in case it is not visible):
[cid:image001.png.01D640BA.A87A7260]
In attempting to solve this issue, we have created undecanal and have processed it through antechamber and parmchk2, in which antechamber applies the gaff force fields. The output files for antechamber and parmchk2 are the prepi and frcmod files, respectively.
The files listed below are loaded into xleap:
Undecanal.prepi
Undecanal.frcmod
We edit the undecanal molecule, remove the hydrogen that is adjacent to the carbonyl in order to prepare the unit for dipeptide linkage. The head and tail of the undecanal group are set accordingly. The ULV monomer is then created:
ULV = sequence {und LEU CVAL}
However, we need an frcmod file which can properly identify the bond that was created between undecanal and the leucine residue, while also keeping the protein force fields intact.
We have tried a few scenarios to resolve this issue:
Scenario #1 (SOP attached to this email):
We created the prepi and frcmod files for ULV via antechamber and parmchk2, respectively. However, this applies the gaff force field parameters to the entire molecule. As mentioned previously, we would like to maintain the protein force fields for the dipeptide head group. Although this allows us to have an frcmod file that sets the correct bond parameters, it does not have the desire force field parameters and gives the entire molecule one residue name as it has applied gaff force field parameters. We need protein force fields for the dipeptide. On the other hand, this runs through the entire simulation.
Scenario #2 (SOP attached to this email):
The mol2 file of ULV was only run through parmchk2 (skipped antechamber to avoid gaff parameters on the dipeptide head group) to obtain the frcmod files of the ULV structure. When loading the frcmod and mol2 file of ULV into xleap, the structure maintained the correct residue names, gaff carbon chain and protein force fields for the dipeptide head group. This allowed us to save the prmtop and inpcrd files for this structure. However, the simulation of the single ULV monomer runs through minimization but once it gets to the warmup, it says “Coordinate resetting (SHAKE) cannot be accomplished, deviation is too large. NITER, NIT, LL, I and J are: 0 2 32 47 50” and does not complete the rest of the simulation.
Comparing Scenarios:
Approach
Pros
Cons
Scenario #1
Runs through simulations
Only gaff force fields
Scenario #2
Protein and gaff force fields
Fails in simulation
New Approach?
In addition to building the ULV unit in Xleap, we have also attempted to build in Avogadro, Molecular Operating Environment (MOE), Pymol and Spartan but we are only able to process these if we run these structures through antechamber, which we do not want.
Another thread in the Amber mailing list with the subject line “Xleap – disappearance of selection when trying to relax selection” was useful in that Dr. David Case alluded that it was possible to “[build]… entirely with gaff. That should(?) get you structures. Then you can input those structures and generate the mixed force fields you want.” Provided with this information, I am not entirely sure as to approach properly mixing force fields in a molecule that has already been entirely created with gaff. I would greatly appreciate any advice on how to resolve this issue.
Best regards,
Mauro A. Garcia
On 6/4/20, 5:56 PM, "Billiot, Eugene" <Eugene.Billiot.tamucc.edu> wrote:
Thanks Carlos, I am now able to create the prmtop and inpcrd files I needed "successfully". At least I hope so. Before I was not able to create these files. It would always give errors in Xleap and they would not be generated.
-----Original Message-----
From: Carlos Simmerling <carlos.simmerling.gmail.com>
Sent: Thursday, June 4, 2020 12:13 PM
To: AMBER Mailing List <amber.ambermd.org>
Subject: Re: [AMBER] help with building an amino acid based surfactant molecule
Carefully check the parmchk2 step, and maybe look at the output it creates
On Thu, Jun 4, 2020, 1:12 PM Billiot, Eugene <Eugene.Billiot.tamucc.edu>
wrote:
> I have tried that before and loaded the frcmod file while I was in
> Xlaep but to be honest I have tired so many things I may not have used
> that correctly. I will try that again and try to follow the steps in
> the TUTORIAL B5 - Simulating the Green Fluorescent Protein using it as
> a guide. Hopefully I can get it to work. Thanks
>
>
> -----Original Message-----
> From: Carlos Simmerling <carlos.simmerling.gmail.com>
> Sent: Thursday, June 4, 2020 11:52 AM
> To: AMBER Mailing List <amber.ambermd.org>
> Subject: Re: [AMBER] help with building an amino acid based surfactant
> molecule
>
> Hi Eugene,
> did you do the parmchk2 step of the GFP tutorial? What did it do? This
> is the part that will try to guess your cross terms.
>
> At this point, we have our residue library containing the charges for
> the atoms in our modified amino acid . We next need to check that the
> covalent parameters (bonds, angles, and dihedrals) that will be needed
> are available. The *parmchk2* program figures out what parameters will
> be needed and checks to see if they are in the standard files. If not,
> it tries to make educated guesses, and puts these new parameters into
> a file we are calling "frcmod.cro" here.
>
>
> On Thu, Jun 4, 2020 at 11:56 AM Carlos Simmerling <
> carlos.simmerling.gmail.com> wrote:
>
> > no - unless you have all upper case (protein force field) or all
> > lower case (gaff), you will need new mixed terms to cross the boundary.
> >
> > On Thu, Jun 4, 2020 at 11:29 AM Billiot, Eugene
> > <Eugene.Billiot.tamucc.edu>
> > wrote:
> >
> >> Carlos,
> >> I am not sure if I understand your suggestion , can I simply change
> >> the gaff atom type names associated with the errors to protein
> >> associated atom type names (upper case) like those associated with
> >> amide bonds in proteins?
> >>
> >> -----Original Message-----
> >> From: Carlos Simmerling <carlos.simmerling.gmail.com>
> >> Sent: Thursday, June 4, 2020 10:22 AM
> >> To: AMBER Mailing List <amber.ambermd.org>
> >> Subject: Re: [AMBER] help with building an amino acid based
> >> surfactant molecule
> >>
> >> Sorry forgot to add thst this is usually done by manually creating
> >> the needed terms, taking the force constants from gafd by analogy
> >> and changing the atom types to those in the protein model (upper case).
> >>
> >> On Thu, Jun 4, 2020, 11:20 AM Carlos Simmerling <
> >> carlos.simmerling.gmail.com>
> >> wrote:
> >>
> >> > It looks like you're missing the cross terms between gaff and the
> >> > protein force field. This would be expected when you combine 2
> >> > force fields in the same molecule, which is what you said you
> >> > want to do so at least part part seems to have gone properly.
> >> > Next you would need to prepare those boundary terms manually, I
> >> > am not aware of a tool to do it (others on the list may have
> >> > tools....). In general combining 2 force fields is a more
> >> > advanced method since they don't know how to
> >> talk to each other at the boundary.
> >> >
> >> > On Thu, Jun 4, 2020, 11:01 AM Billiot, Eugene
> >> > <Eugene.Billiot.tamucc.edu>
> >> > wrote:
> >> >
> >> >> Hello Folks,
> >> >> I have been struggling for over a month to build an
> >> >> amino acid-based lipid with undecanal as the hydrophobic tail
> >> >> and the carbon on the C=O group of the undecanal forming an
> >> >> amide bond with the first amino acid in my dipeptide based
> >> >> surfactant. When I run antechamber on my surfactant molecule,
> >> >> it replaces amino acid force field parameters with gaff
> >> >> forcefield parameters and it converts all of the residues (such
> >> >> as
> >> >> und-LEU-VAL) with und being the undecanal group to one residue
> >> >> (the first one - und) losing the amino acid residue information.
> >> >> I do not want to use gaff forcefield parameters for the
> >> >> amino acid head groups because I suspect the protein database
> >> >> forcefields are better descriptors. How can I build the
> >> >> surfactant molecule with three residue names such as described
> >> >> above (und-LEU-VAL) with und described by the gaff forcefield
> >> >> parameters and the amino acid moieties described by the protein
> >> >> forcefield parameters? Every time I build the sequence in Xleap
> >> >> and try to run "saveamberparm" I get the errors below which are
> >> >> related to the amide linkage between the C=O on the undecanal
> >> >> forming an amide bond with the first amino acid in my dipeptide
> based surfactant (Leucine in the example I mentioned).
> >> >> The problem is that I am somewhat of novice in terms of
> >> >> computational chemistry. The one Amber tutorial that I found
> >> >> most useful is TUTORIAL B5 - Simulating the Green Fluorescent Protein.
> >> >> I feel confident that the answer to my problem lies in that
> >> >> tutorial, but I get lost in trying to understand what the most
> >> >> relevant points are in helping me address my problem. Any help
> >> >> would be greatly
> >> appreciated. Thanks!!!!!
> >> >>
> >> >> NOTE: In case it is not clear, when I create my
> >> >> undecanal-LEU-VAL sequence in Xleap, I named it ULV, thus the
> >> >> three-letter abbreviation
> >> >> (ULV) below.
> >> >> > saveamberparm ULV ULV.prmtop ULV.inpcrd
> >> >> Checking Unit.
> >> >>
> >> >> Warning: The unperturbed charge of the unit (-1.000000) is not zero.
> >> >>
> >> >> Note: Ignoring the warning from Unit Checking.
> >> >>
> >> >> Building topology.
> >> >> Building atom parameters.
> >> >> Building bond parameters.
> >> >>
> >> >> Error: Could not find bond parameter for: c - N Building angle
> >> >> parameters.
> >> >>
> >> >> Error: Could not find angle parameter: o - c - N
> >> >>
> >> >> Error: Could not find angle parameter: c - N - H
> >> >>
> >> >> Error: Could not find angle parameter: c - N - CX
> >> >>
> >> >> Error: Could not find angle parameter: c3 - c - N Building
> >> >> proper torsion parameters.
> >> >>
> >> >> Error: ** No torsion terms for o-c-N-H
> >> >>
> >> >> Error: ** No torsion terms for o-c-N-CX
> >> >>
> >> >> Error: ** No torsion terms for c3-c-N-H
> >> >>
> >> >> Error: ** No torsion terms for c3-c-N-CX Building improper
> >> >> torsion parameters.
> >> >> old PREP-specified impropers:
> >> >> <und 1>: C10 H34 C11 O12
> >> >> total 5 improper torsions applied
> >> >> 1 improper torsions in old prep form Building H-Bond parameters.
> >> >> Incorporating Non-Bonded adjustments.
> >> >>
> >> >> Warning: Parameter file was not saved.
> >> >>
> >> >>
> >> >> _______________________________________________
> >> >> AMBER mailing list
> >> >> AMBER.ambermd.org
> >> >> http://lists.ambermd.org/mailman/listinfo/amber
> >> >>
> >> >
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image001.png)
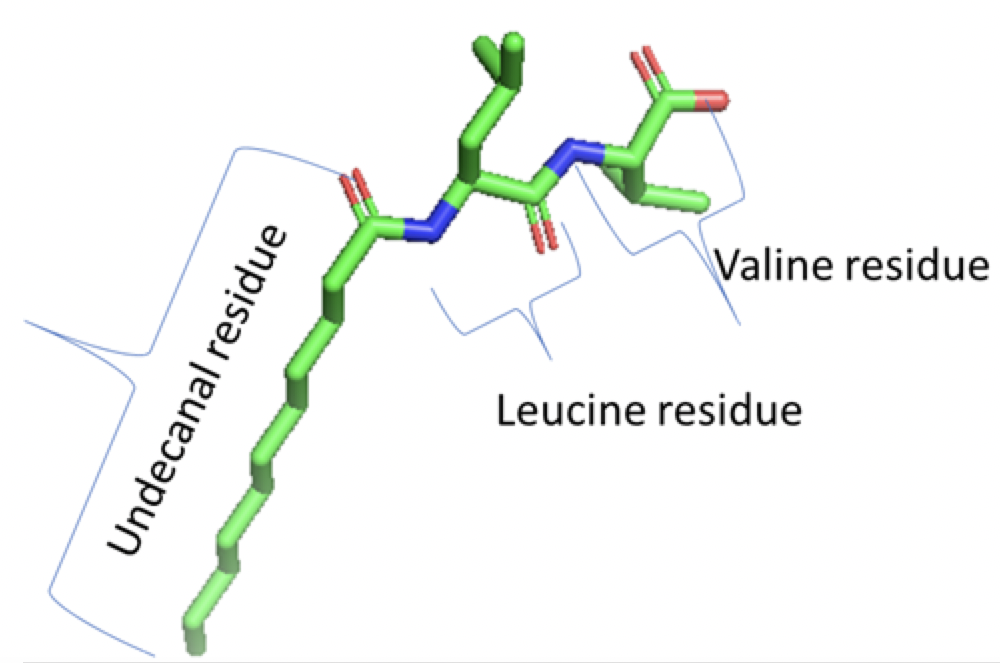
(image/png attachment: ULV.png)
- application/vnd.openxmlformats-officedocument.wordprocessingml.document attachment: Scenario 1.docx
- application/vnd.openxmlformats-officedocument.wordprocessingml.document attachment: Scenario 2.docx
