Date: Fri, 12 Jun 2020 13:24:20 -0400
Hi Amber Comunity,
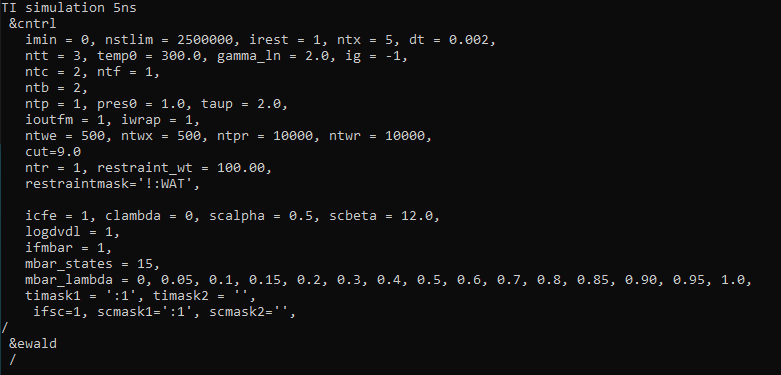
I am using Amber20 TI ( PMEMD.cuda) to calculate benzene's solvation free
energy.
The command for the production run:
*pmemd.cuda -i ti.in <http://ti.in/> -c eq.rst7 -ref eq.rst7 -p ti.prmtop
-O -o ti001.out -inf ti001.info <http://ti001.info/> -e energy.en -r
ti001.rst7 -x ti001.nc <http://ti001.nc/> -l ti001.log*
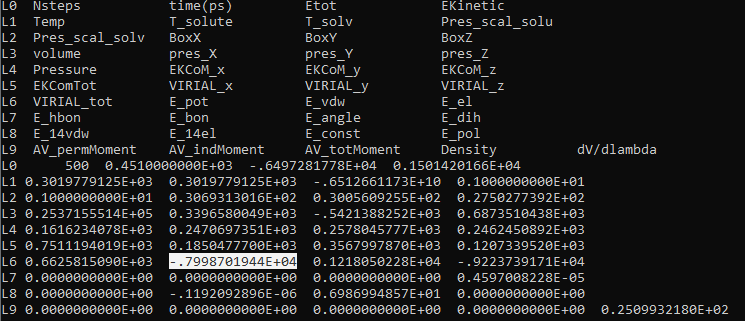
In the generated energy.en file (attached as energy.PNG), does the
Epot value (Highlighted at line L6) include the interaction energy among
the "disappearing" atoms of benzene?
The reason why I ask it is I want to know in Amber, is it correct to
calculate the Enthalpy Change(dH) of benzene solvation process by: dH =
Epot (lambda=0, when benzene in water) - Epot (lambda=1, when benzene
disappeared) if I run this two lambda windows long enough(such as 100 ns).
I appreciate your help in advance!
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ti.PNG)
(image/png attachment: energy.PNG)
