Date: Sat, 20 Feb 2021 19:17:14 +0000
Dear Amber Users and Developers,
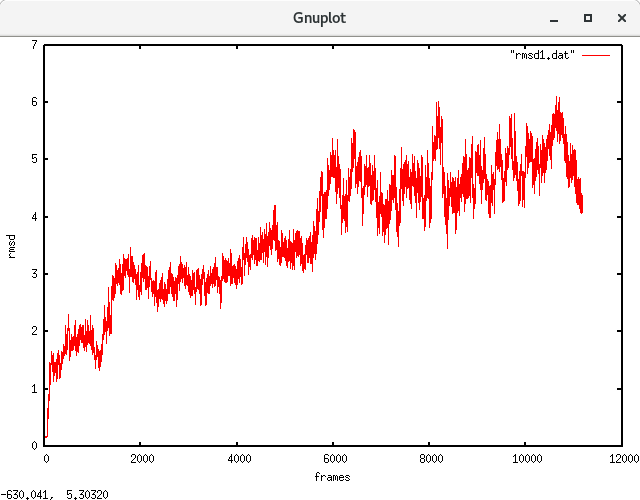
Here I have attached is a rmsd plot obtained for a ~100 ns equilibrated trajectory (saved at every 10 ps) of a DNA/ligand system. I am attempting to calculate absolute binding free energy through a thermodynamic integration (TI) approach. I was hoping if you could clarify to me whether the system is equilibrated enough to proceed with the TI calculations. I was under doubt due to the fluctuations shown throughout the last 6000 frames (60 ns). I understand there isn't an exact answer to this. But I appreciate your opinion on this.
[cid:f8200194-9003-4107-accf-086dd0f15353]
Thank you
Senal Liyanage
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: rmsd-equil100ns.png)
