Date: Wed, 24 Jun 2020 18:35:38 +0000
Hi All
I am a bit confused with regard to the NVT production runs of TI setup I had established a few weeks back.
My ligand of interest is ethanol and my prod.in file looks like this:
[SITE1]$ cat ./0.11505/prod_NVT.in
&cntrl
imin = 0, nstlim = 20000000, dt = 0.001,
irest = 1, ntx = 5, ig = -1,
tempi = 300.0, temp0 = 300.0,
ntwx = 10000, ntwe = 10000, ntwr = 10000, ntpr = 10000,ntwv=-1, ntave =1000,
cut = 11.0,
ntt =3, ntb = 1, ntp = 0, gamma_ln=3, iwrap = 1,
ntc = 1, ntf = 1, tol = 0.00001,
nsnb = 10, nscm = 10,
ioutfm=1,
taup = 2,
ntr=1, restraintmask=':EOH', restraint_wt=10.00,
icfe = 1, clambda = 0.11505, ifsc=1,
timask1=':EOH',timask2='',
scmask1=':EOH', scmask2=''
&end
&ewald
skinnb=2, nfft1=96, nfft2=96, nfft3=96,
/
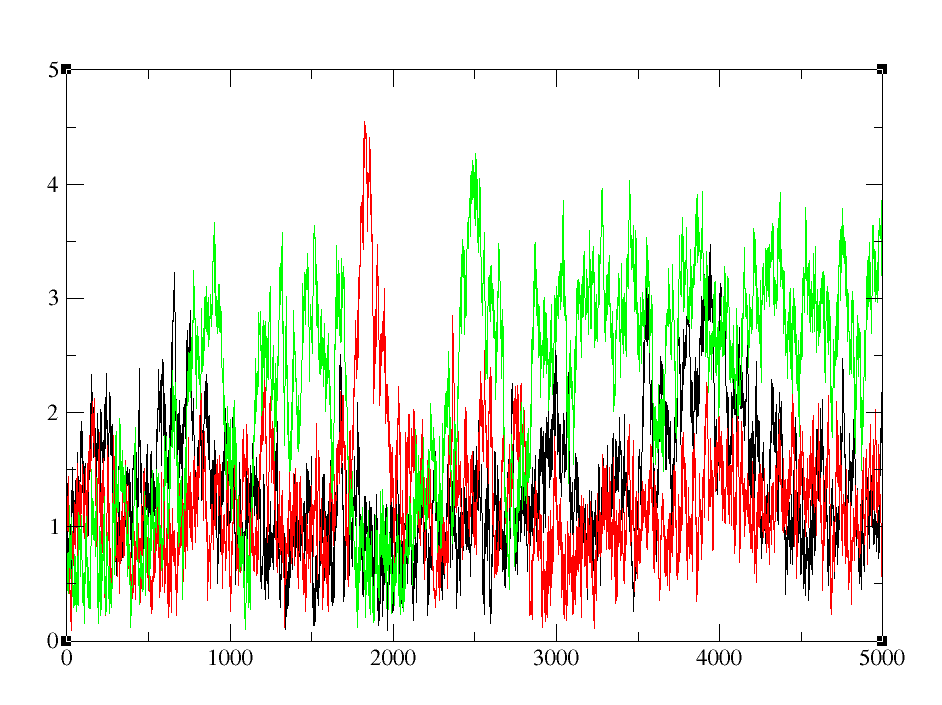
So, after the run was complete, I was curious to see how far my ligand has translated from its tether point.. and after doing a COM distance cpptraj script run, I find that for 3 lambda values 0.11505, 0.20634, and 0.31608; the ligand has moved around quite a bit as in > 3 Angstroms from its initial solvated state. Is this expected or am I doing something wrong in putting the restraint weight and mask?
I thought that 10 kcal/mol is quite a big force to keep ligand restrained in the pocket. I was not expecting a 3-4 Angstrom jiggling of the ligand atoms.
Should I be worried?
[cid:image001.png.01D64A34.03736FA0]
Sent from Mail<https://go.microsoft.com/fwlink/?LinkId=550986> for Windows 10
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 69325509881B4669B6C8002DBD64F67B.png)
