Date: Fri, 12 Dec 2014 14:19:20 +0530
Dear Amber users,
I am new to the MD simulation. I have faced a strange problem in doing
simulation of protein-DNA complex. The problem, questions and the setup
details are given below:
*****PROBLEM********
---------------------------------
I have planned to run 100ns simulation and divide the whole run in 50
restart (each of 2ns). My simulation ran well upto 94ns but at the begining
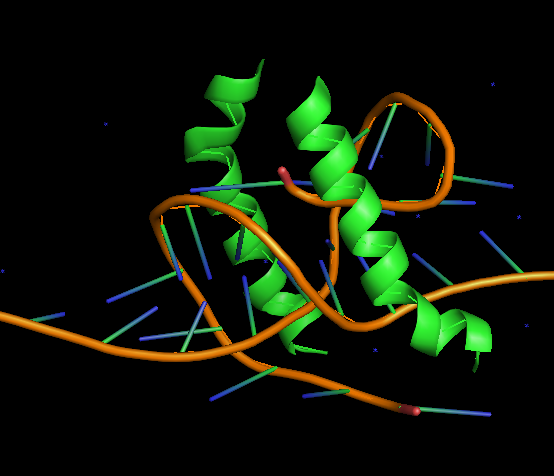
of the next restart I found the DNA get horribly denatured. I have attached
the picture with this mail.
*****QUESTION********
---------------------------------
1. I have checked the prmtop file with rdparm to make sure about the use of
proper force field. But I have doubt whether my way of selection of force
field is correct or not? and also regarding the production run script.
2. I did not get why the DNA get unstable and how can I analyze the cause
and correct it?
*****SETUP********
---------------------------
1. The structure is a crystal structure with all crystal water. DNA residue
no: 103-138
*2.Force field used: ff99SB-ILDN along with parambsc0*
*[tleap file command: source leaprc.ff99SBildn*
* loadoff DNA_CI.lib*
* loadamberparams frcmod.parmbsc0*
*I do did energy minimization (with and without position restraint, heating
(with position restraint), NPT equilibration (gradually relaxing restraint)
and finally performed production run*
*3.Production run:*
&cntrl
imin = 0, irest = 1, ntx = 5,
ntb = 2, pres0 = 1.0, ntp = 1,
taup = 2.0,
cut = 8.0, ntr = 0,
ntc = 2, ntf = 2,
tempi = 300.0, temp0 = 300.0,
ntt = 3, gamma_ln = 1.0, ig = -1, iwrap = 1,
nstlim = 1000000, dt = 0.002, nmropt =1,
ntpr = 2500, ntwx = 2500, ntwr = 2500,
ntwv = 2500, ntwe = 2500
/
&wt type='DUMPFREQ', istep1=2000
/
&wt type='END'
/
LISTOUT=POUT
DISANG=DIST.rst
DUMPAVE=rest_values_INPRORUN1
END
*4. Distance restraint input file*
2 ALA O 6 ALA N 3.0
3 LEU O 7 ARG N 3.0
53 ALA O 57 ALA N 3.0
54 LEU O 58 ARG N 3.0
50 LEU N 46 ARG O 2.9
49 LYS N 45 ALA O 3.2
101 LEU N 97 ARG O 3.0
100 LYS N 96 ALA O 3.0
103 DG5 O6 138 DC3 H41 2.0
103 DG5 H1 138 DC3 N3 1.9
103 DG5 H22 138 DC3 O2 3.5
121 DG5 O6 120 DC3 H41 1.9
121 DG5 H1 120 DC3 N3 1.9
121 DG5 H22 120 DC3 O2 3.5
*5. Commands used:*
to generate DIST.rst file:
****************************
makeDIST_RST -upb DNA_Protein-endDISTRST -pdb emin_NoRST.pdb -rst DIST.rst
For production run:
**********************
nohup mpiexec.hydra -n 8 sander.MPI -O -i production1.in -o prodrun1.out -p
GCN4_CREB_inbox.prmtop -c equil_NPT.rst -r prodrun1.rst -x prodrun1.mdcrd
nohup mpiexec.hydra -n 8 pmemd.MPI -O -i production2.in -o prodrun2.out -p
GCN4_CREB_inbox.prmtop -c prodrun1.rst -r prodrun2.rst -x prodrun2.mdcrd
*NOTE: I was initially unsure about using "pmemd" that's why I started
first run with sander and after reading maual and Amber blog, I started
using "pmemd".
Looking forward for your reply.
regards
Aditya
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2014-12-12_03:14:01.png)
