Date: Wed, 2 Nov 2022 20:39:10 +0000
Hi Anselm,
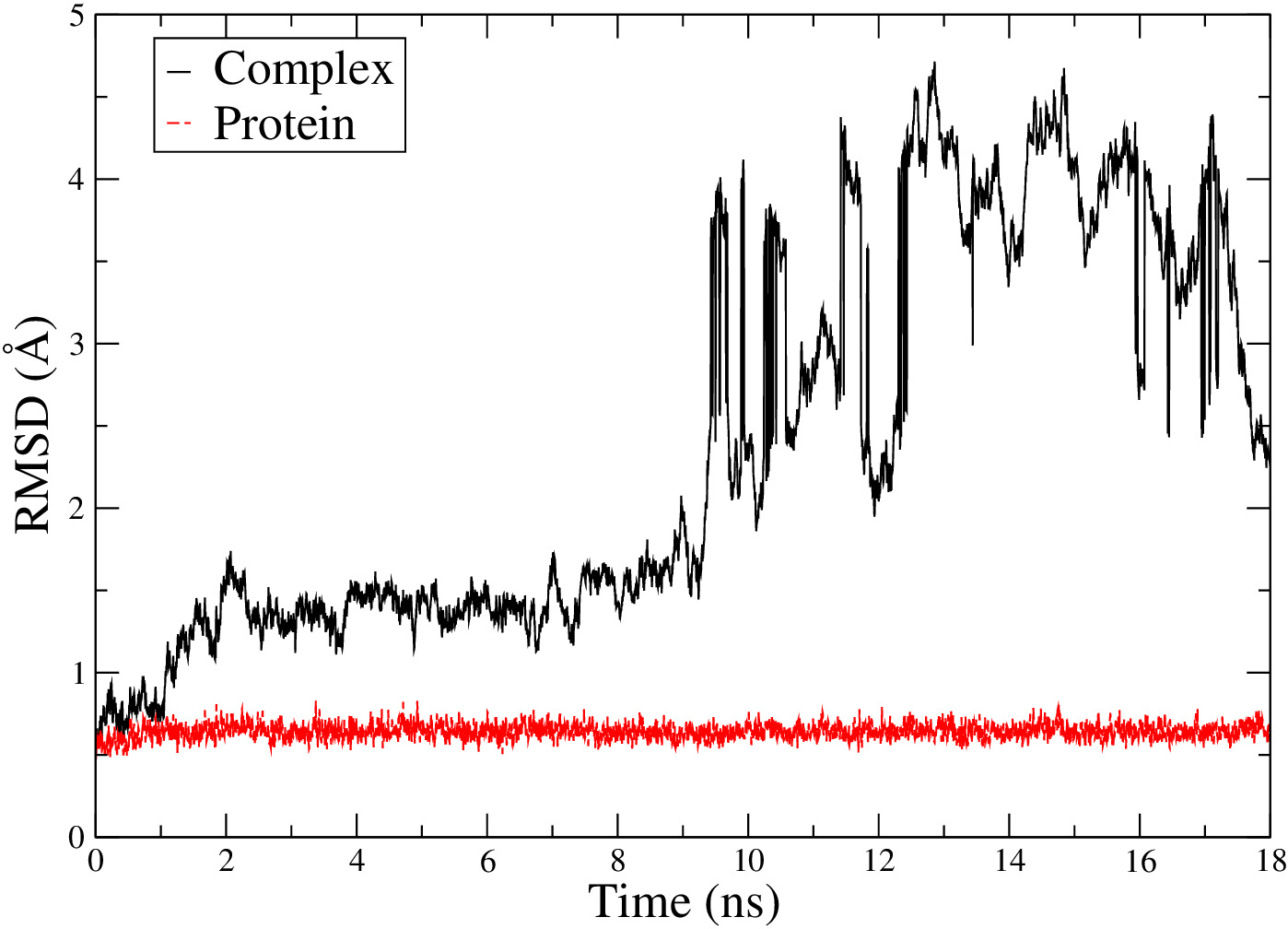
Thank you very much for the reply, which is very helpful. Following your instruction, I unwrapped the trajectories using autoimage. Most of the trajectories were fixed except one, which still showed jumping as the RMSD result figure shown below. I am wondering what is the reason for it, and how to fix it. Thank you!
[cid:60f78aeb-9137-455c-851f-da1dece9b128]
Best,
Laura
________________________________
From: Dr. Anselm Horn via AMBER <amber.ambermd.org>
Sent: Wednesday, November 2, 2022 11:41
To: amber.ambermd.org <amber.ambermd.org>
Subject: Re: [AMBER] about mmgbsa method
Laura,
if your organic ligand left its binding pocket during the simulation,
this is no "PBC effect", but the result of your simulation. (If this is
unexpected or not in accordance with experimental findings, maybe your
setup or your ligand parameters are not right.)
Perhaps you could use cpptraj to unwrap or autoimage your trajectory.
If VMD displays "broken" bonds in a structure, it may just be a
visualization artifact, because the distance between the two atoms is
larger than the program-specific built-in cutoff for a bond. This does
not mean, that the bond broke and maybe formed again.
Concerning a MMPBSA analysis:
This post-production analysis makes only sense for a valid trajectory,
i.e. when the ligand stays intact and bound to the protein.
Maybe this helps.
Best,
Anselm
Bioinformatik | NHR.FAU
Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU
Germany
Am 02.11.2022 um 16:19 schrieb laura zhang via AMBER:
> Dear all,
>
> I did AMBER simulations on a protein binding with a small organic molecule. Checking the trajectory, I can see that the small organic compounds had PBC effect that it departured from the protein. However, when I used pbc unwrap in vmd program to unwrap the trajectory, the small compound side chains broke here and there. I am wondering how I can unwrap the amber trajectory correctly.
> Another question is, under such kind of condition, when I did mmgbsa binding energy calculation on the protein binding with the small organic molecule, will the result be reliable? what should I do in order to get reliable binding energy result? Thank you very much.
>
> Laura
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> https://nam12.safelinks.protection.outlook.com/?url=http%3A%2F%2Flists.ambermd.org%2Fmailman%2Flistinfo%2Famber&data=05%7C01%7C%7C659dc48e107a42c44d6e08dabce8b8e4%7C84df9e7fe9f640afb435aaaaaaaaaaaa%7C1%7C0%7C638030004983113254%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=8ObE6yRbv9Dac%2F9YIRHbdmsGYvI77Ov5OqfkeGfhELQ%3D&reserved=0
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
https://nam12.safelinks.protection.outlook.com/?url=http%3A%2F%2Flists.ambermd.org%2Fmailman%2Flistinfo%2Famber&data=05%7C01%7C%7C659dc48e107a42c44d6e08dabce8b8e4%7C84df9e7fe9f640afb435aaaaaaaaaaaa%7C1%7C0%7C638030004983113254%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=8ObE6yRbv9Dac%2F9YIRHbdmsGYvI77Ov5OqfkeGfhELQ%3D&reserved=0
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: image.png)
