Date: Thu, 16 Dec 2021 01:22:43 +0000
To whom it may concern
Hope this email finds you well
Currently I am working on the building the parameters files with AMBER for reductive dehalogenase PceA.
Within my protein, there is an organometallic complex cobalt corrinoid.
By using the published reference, I could construct the lib file, but not for the frcmod file.
The xleap keeps reporting the error message for no torsion terms error even though I specified them in frcmod file, as attached screen shot and frcmod input
Similar to the problem posted as the following link
http://archive.ambermd.org/201711/0062.html
May you please kindly help me to identify problem and any instructions to solve it? Much appreciated
Yi Ren
0420248895
PhD candidate UNSW
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
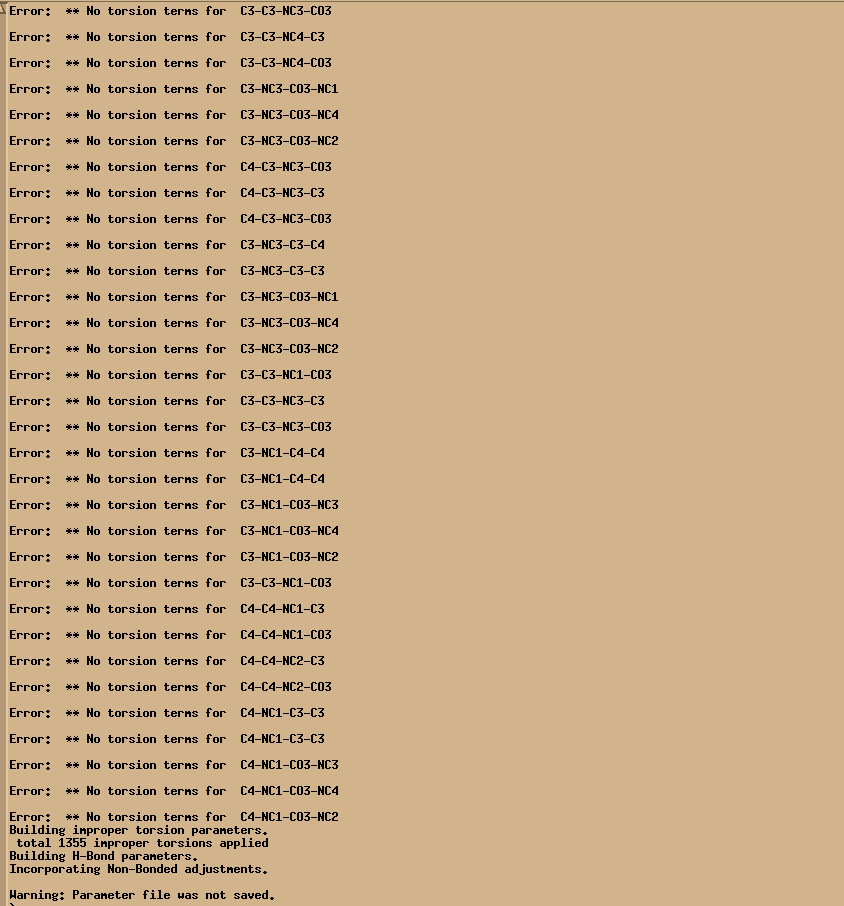
(image/png attachment: error_no_torsion_terms.png)
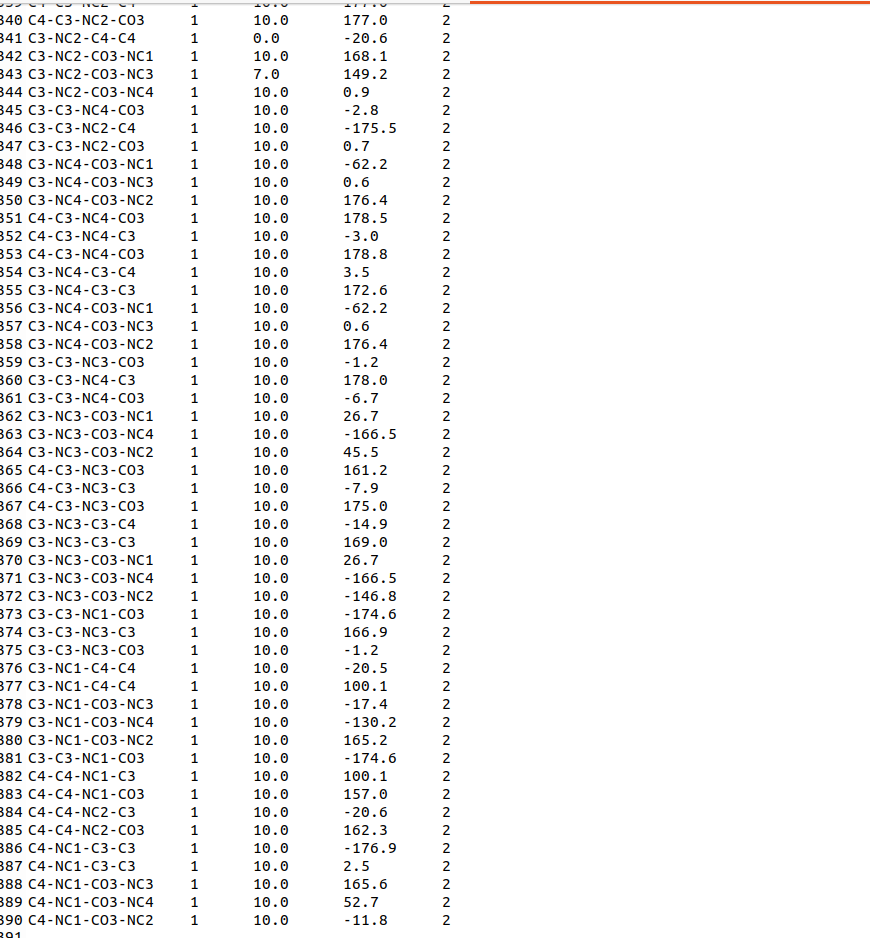
(image/png attachment: frcmod_input.png)
