Date: Wed, 6 Oct 2021 23:38:09 +0000
Hi All,
I am simulating a dimer protein with water molecules. Initially I am doing restrained energy minimization, after that I am minimizing the energy of whole system. I am heating the system by restraining the protein upto 100 K. When I analyzed my trajectory file of the heating . I am getting voids symmetrical to box dimension. Attached is the image of whole system minimization and heating run. Can anyone help me, Why I am getting those symmetrical voids during the heating stage?
Here are my script files that I am using-
Restrained Minimization
&cntrl
imin=1, maxcyc=10000, ncyc=5000,
ntr=1, ntxo=1, ntb=1,
cut=10.0
&end
Group input for restrained atoms
10.0
RES 1 482
END
END
Whole System Minimization
&cntrl
imin=1,
maxcyc=6000, ncyc=2000,
ntb=1, ntr=0,
&end
Heating Stage-
&cntrl
imin=0, irest=0, ntx=1, ntb=1,
cut=10.0, ntr=1, ntc=2, ntf=2,
tempi=0.0,
temp0=100.0,
ntt=3,
gamma_ln=1,
nstlim=2500, dt=0.002
ntpr=100, ntwx=100, ntwr=1000
/
Keep protein fix
10.0
RES 1 482
END
END
trajin file
trajin heat_V_md1.nc
trajout heat_V_md1_frames.pdb
#command to run: cpptraj 1QWT_D_W.top < extract_frames.trajin > heat_V_md1_frames.out
Regards,
Hemant Nagar
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
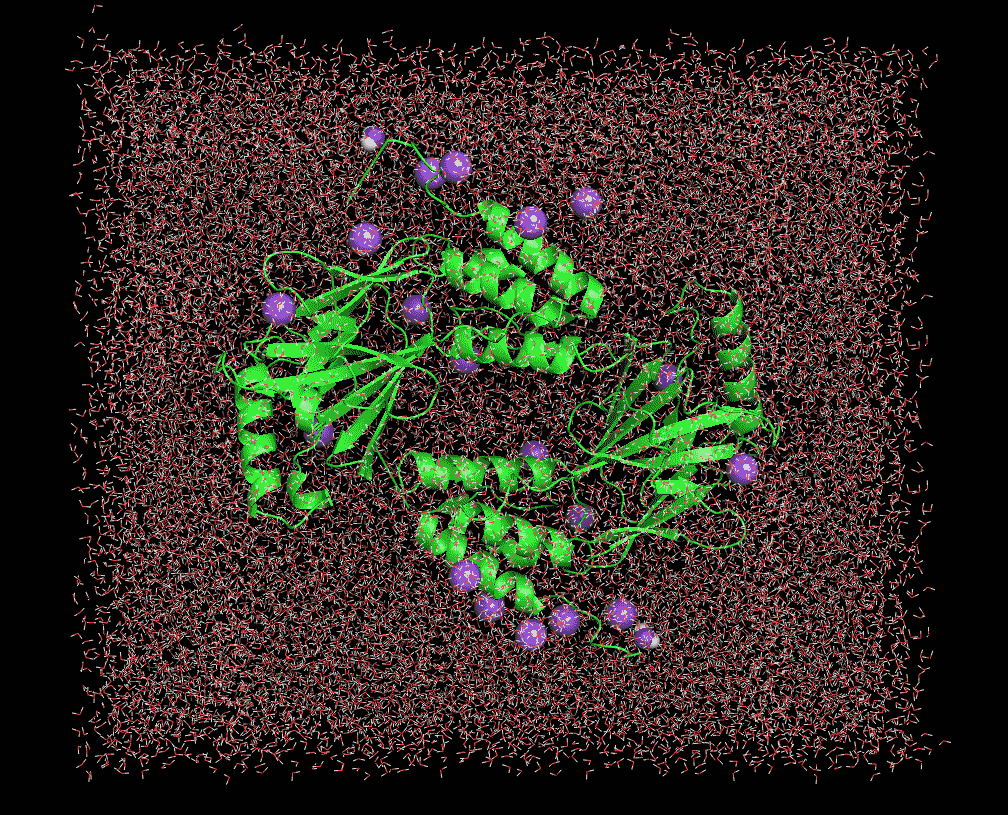
(image/png attachment: min_wholesystem1.PNG)
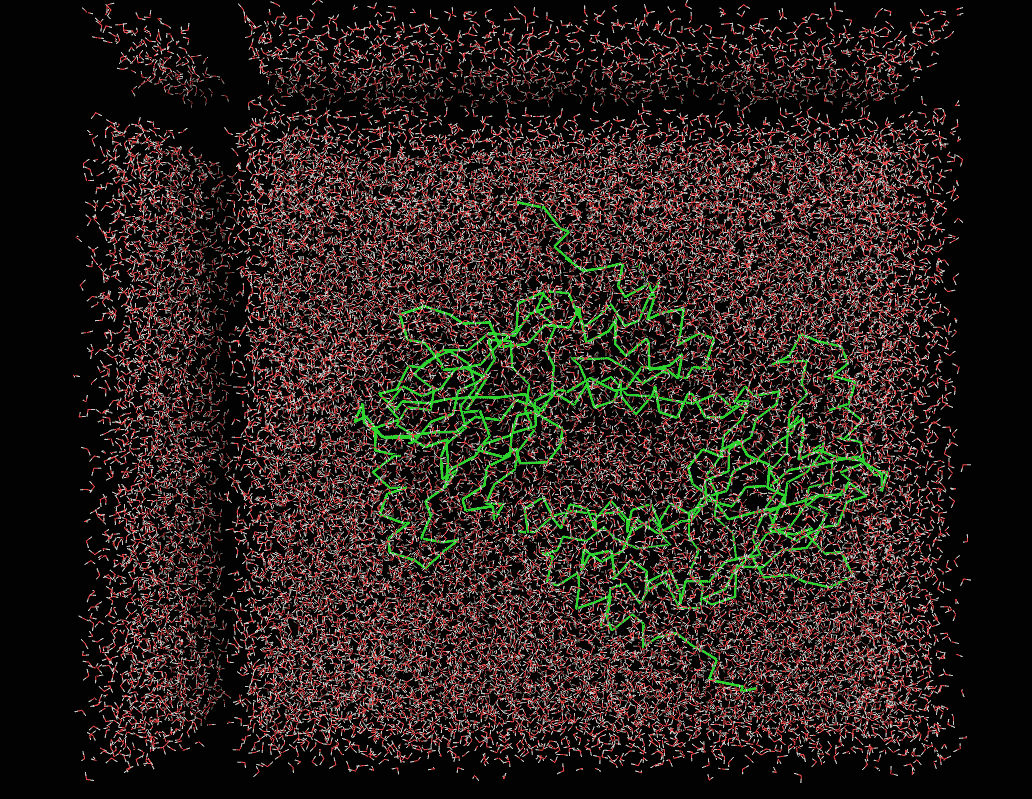
(image/png attachment: heat_V_.PNG)
