Date: Thu, 4 Jun 2020 18:34:26 +0300
Hello,
I am trying to perform molecular dynamics simulation for DNA-ligand complex.
For this, I used this tutorials and files
https://ambermd.org/tutorials/basic/tutorial1/section5.htm
all minimization, heating and production files are the same with this
tutorial. But instead of tutorial's DNA I used 1BNA PDB code DNA and I u
sed my ligand as well.
After I docked my ligand and 1BNA, I made my DNA-ligand complex and then I
used gaff and my novel force field frcmod file for the ligand after I
solved my complex in TIP3P water I followed the instruction the tutorial I
mentioned above.
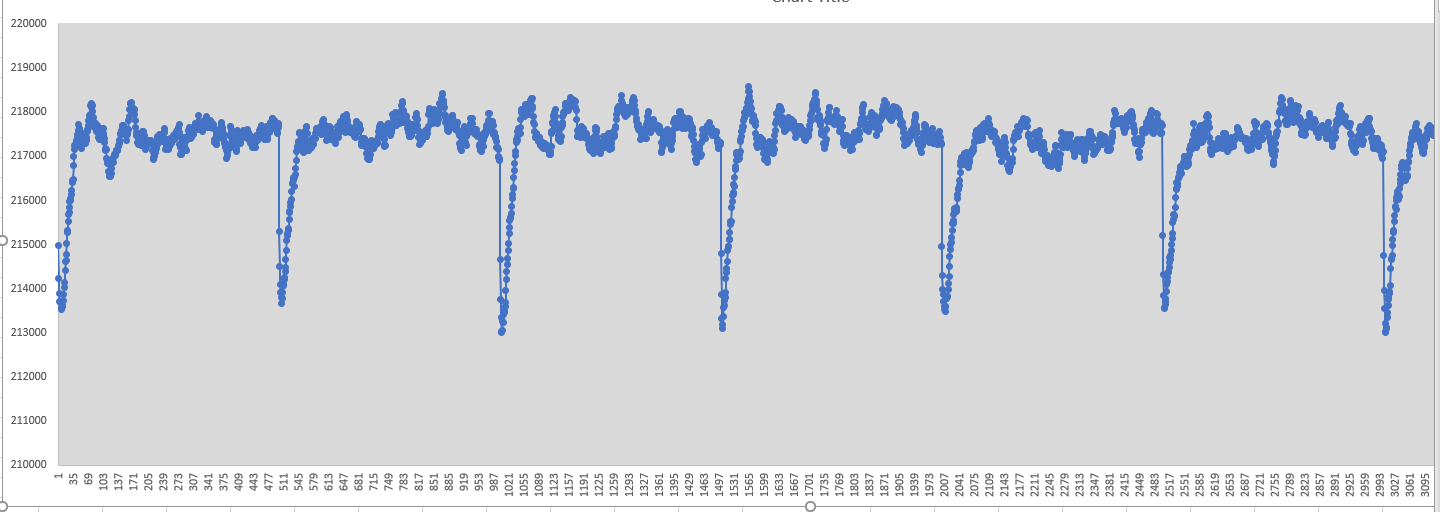
Result was very surprising here is my volume chart
(if it does not seem chart is here: https://ibb.co/TRkzVxh)
[image: image.png]
and my kinetic and potential energy charts are similar. in the beginning
of every 500 steps, volume and energy is dropping suddenly then everything
going up normal. My computational power is not big so I couldn't try
another method. My questions are
1) What is happening in the beginning of every 500 steps?
2) Is this normal or something went wrong?
3) if it is wrong how can I solve this?
Thank you
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
