Date: Wed, 27 Mar 2019 11:32:49 +0530
I tried PM6 without any restraints as Andreas suggested. While there was no
large changes in temperature (298-304K). But it gave some unrealistic
results.
Hydrogen attached to terminal Nitrogen of Arginine (in QM region) flew off
(distance plot attached) and it (Nitrogen) abstracted a Hydrogen from water
(plot attached). I don't think this could be the case in vivo.
I also started unrestrained QMMM with gaussian (as others sugested) but it
will take few days to get to complete 1 ps run.
I would also like to add that i have already done some equilibriation on
this system using first HF and then varying basis sets with B3LYP and it
worked fine till now.
On Wed, Mar 27, 2019 at 7:51 AM Abhilash J <md.scfbio.gmail.com> wrote:
> As everyone suggested restraints might be the cause, I am retrying with
> the restraints removed. It will take few days to get near 800 ps as it is a
> large QM region.
> I will also try using semiempirical Hamiltonian PM6 and hope that it
> helps.
>
> On Wed, Mar 27, 2019 at 3:24 AM Goetz, Andreas <agoetz.sdsc.edu> wrote:
>
>> The Gaussian output has an entry “Atoms too close”. This means that part
>> of your QM region has a bad geometry. Given that your first few hundred
>> steps were stable, this must have happened at some point during your
>> trajectory, perhaps because of your restraints. As a consequence, Gaussian
>> is probably not able to optimize the correct ground state electron density,
>> which results in bad forces both for the QM region and the classical atoms
>> that are coupled via the QM/MM Hamiltonian (within your QM cutoff, as you
>> observe).
>>
>> You have to be careful equilibrating QM/MM calculations. Perhaps try a
>> semiempirical Hamiltonian like PM6 first to see how the system behaves.
>>
>> All the best,
>> Andy
>>
>> —
>> Dr. Andreas W. Goetz
>> Assistant Research Scientist
>> San Diego Supercomputer Center
>> Tel: +1-858-822-4771
>> Email: agoetz.sdsc.edu
>> Web: www.awgoetz.de
>>
>> > On Mar 26, 2019, at 6:33 AM, Abhilash J <md.scfbio.gmail.com> wrote:
>> >
>> > Yes, The temperature is increasing too rapidly. I did not notice that
>> > earlier. Thanks for pointing it out. But it was stable till 0.796 ps.
>> > I don't know what can caused the thermostat to fail. It did not fail in
>> the
>> > last run of (1 ps) the same complex (that too was QM).
>> > One interesting point is that the erratic movement of atoms happens in
>> the
>> > region described in the 10A cut sphere region of QM. Rest of the protein
>> > atoms are fine.
>> > So I think it might be linked to QM part.
>> >
>> > This is the active site containing domain part of a larger protein. I
>> have
>> > run over 500 ns with the complete protein, and it runs fine using MM.
>> > Current complex (active site domain part) has some active site residues,
>> > ATP, MG2+, K+ ions and catalytic water in QM region. Rest of the
>> protein,
>> > water and ions are treated as MM.
>> > One thing i can try is to remove the restraints on the initial amino
>> acids
>> > of the protein and try this again. I will try this.
>> > I think i should point out that I am using sander with Gaussian
>> interface.
>> > In case it helps.
>> >
>> >
>> >
>> > On Tue, Mar 26, 2019 at 4:09 PM Bill Ross <ross.cgl.ucsf.edu> wrote:
>> >
>> >> How does it go without QMM?
>> >>
>> >> Bill
>> >>
>> >>
>> >> On 3/26/19 2:45 AM, Szymon Żaczek wrote:
>> >>> Dear Abhilash,
>> >>>
>> >>> please, have a look at temperatures in your system: Starting from
>> >>> 798th step onwards it is rising very rapidly., much above the defined
>> >>> 300 K. This may be caused by a plethora of issues but generally it
>> >>> means that your system is unstable. Restraint energy in your system is
>> >>> pretty high comparing to the restraint value that you defined - it may
>> >>> be the main cause of your system's instability.
>> >>>
>> >>> Kind regards,
>> >>> Szymon
>> >>>
>> >>> wt., 26 mar 2019 o 10:27 Abhilash J <md.scfbio.gmail.com> napisał(a):
>> >>>> Hi everyone,
>> >>>>
>> >>>> I am trying QMMM for a protein ATP complex. After 1.798 ps the
>> >> system
>> >>>> suddenly shows vmax error
>> >>>> vlimit exceeded for step 798; vmax = **********
>> >>>>
>> >>>> When i googled the error i found that it occurs when there is atom
>> >>>> overlap. So i checked the trajectory. The atoms in the region around
>> >> the QM
>> >>>> region (seemingly 10A, probably due to cut variable) suddenly move
>> away
>> >>>> from each other. And Gaussian shows error (attached below)
>> >>>> I cannot understand why this is so.
>> >>>> * The AMBER output file look like this:*
>> >>>>
>> =======================================================================
>> >>>> NSTEP = 796 TIME(PS) = 0.796 TEMP(K) = 283.32
>> PRESS =
>> >>>> 0.0
>> >>>> Etot = -2958635.1773 EKtot = 35534.3457 EPtot =
>> >>>> -2994169.5230
>> >>>> BOND = 23904.6935 ANGLE = 2717.0388 DIHED =
>> >>>> 4447.2846
>> >>>> 1-4 NB = 1264.0951 1-4 EEL = 14929.0965 VDWAALS =
>> >>>> 20957.1806
>> >>>> EELEC = -192019.8681 EHBOND = 0.0000 RESTRAINT =
>> >>>> 61.9493
>> >>>> EXTERNESCF= -2870430.9932
>> >>>> EAMBER (non-restraint) = -2994231.4723
>> >>>> Ewald error estimate: 0.6943E-04
>> >>>>
>> >>
>> ------------------------------------------------------------------------------
>> >>>>
>> >>>>
>> >>>> NSTEP = 797 TIME(PS) = 0.797 TEMP(K) = 286.04
>> PRESS =
>> >>>> 0.0
>> >>>> Etot = -2958784.2270 EKtot = 35875.5721 EPtot =
>> >>>> -2994659.7991
>> >>>> BOND = 23435.8580 ANGLE = 2718.9276 DIHED =
>> >>>> 4448.9569
>> >>>> 1-4 NB = 1265.2528 1-4 EEL = 14927.2921 VDWAALS =
>> >>>> 20954.0672
>> >>>> EELEC = -192037.1185 EHBOND = 0.0000 RESTRAINT =
>> >>>> 62.1595
>> >>>> EXTERNESCF= -2870435.1946
>> >>>> EAMBER (non-restraint) = -2994721.9586
>> >>>> Ewald error estimate: 0.5550E-04
>> >>>>
>> >>
>> ------------------------------------------------------------------------------
>> >>>>
>> >>>> vlimit exceeded for step 797; vmax = 223.1064
>> >>>>
>> >>>> NSTEP = 798 TIME(PS) = 0.798 TEMP(K) = 692.97
>> PRESS =
>> >>>> 0.0
>> >>>> Etot = -2907774.1888 EKtot = 86911.7535 EPtot =
>> >>>> -2994685.9423
>> >>>> BOND = 23514.7837 ANGLE = 2718.7990 DIHED =
>> >>>> 4450.6613
>> >>>> 1-4 NB = 1265.9952 1-4 EEL = 14925.6770 VDWAALS =
>> >>>> 20952.9524
>> >>>> EELEC = -192104.7780 EHBOND = 0.0000 RESTRAINT =
>> >>>> 62.3920
>> >>>> EXTERNESCF= -2870472.4249
>> >>>> EAMBER (non-restraint) = -2994748.3344
>> >>>> Ewald error estimate: 0.6306E-04
>> >>>>
>> >>
>> ------------------------------------------------------------------------------
>> >>>>
>> >>>> *vlimit exceeded for step 798; vmax = ***********
>> >>>>
>> >>>> NSTEP = 799 TIME(PS) = 0.799 TEMP(K) = 9298.03
>> PRESS =
>> >>>> 0.0
>> >>>> Etot = -1820708.4883 EKtot = 1166155.2961 EPtot =
>> >>>> -2986863.7844
>> >>>> BOND = 29386.9020 ANGLE = 3033.3225 DIHED =
>> >>>> 4524.6387
>> >>>> 1-4 NB = 1281.3174 1-4 EEL = 14909.3089 VDWAALS =
>> >>>> 22456.8078
>> >>>> EELEC = -192281.1708 EHBOND = 0.0000 RESTRAINT =
>> >>>> 73.8498
>> >>>> EXTERNESCF= -2870248.7608
>> >>>> EAMBER (non-restraint) = -2986937.6342
>> >>>> Ewald error estimate: 0.7918E-04
>> >>>>
>> >>
>> ------------------------------------------------------------------------------
>> >>>> ...
>> >>>> ...
>> >>>> ...
>> >>>>
>> >>
>> ------------------------------------------------------------------------------
>> >>>>
>> >>>> vlimit exceeded for step 837; vmax = **********
>> >>>>
>> >>>> NSTEP = 838 TIME(PS) = 0.838 TEMP(K) = 48220.98
>> PRESS =
>> >>>> 0.0
>> >>>> Etot = ************** EKtot = 6047856.4596 EPtot =
>> >>>> **************
>> >>>> BOND = 14241797.7566 ANGLE = 271885.7319 DIHED =
>> >>>> 13378.3660
>> >>>> 1-4 NB = ************** 1-4 EEL = 12495.8527 VDWAALS =
>> >>>> **************
>> >>>> EELEC = -184401.7970 EHBOND = 0.0000 RESTRAINT =
>> >>>> 28859.2744
>> >>>> EXTERNESCF= -2789848.0401
>> >>>> EAMBER (non-restraint) = **************
>> >>>> Ewald error estimate: 0.5005E-04
>> >>>>
>> >>
>> ------------------------------------------------------------------------------
>> >>>>
>> =======================================================================
>> >>>>
>> >>>>
>> >>>>
>> >>>> *Gaussian error:*
>> >>>>
>> ========================================================================
>> >>>> Leave Link 101 at Tue Mar 26 08:37:13 2019, MaxMem=10737418240 cpu:
>> >>>> 13.4
>> >>>> (Enter /scf-data/apps/gaussian/g09/l103.exe)
>> >>>>
>> >>>>
>> >>
>> GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
>> >>>> Berny optimization.
>> >>>> Initialization pass.
>> >>>> Trust Radius=3.00D-01 FncErr=1.00D-07 GrdErr=1.00D-07
>> >>>> Number of steps in this run= 2 maximum allowed number of steps=
>> >>>> 2.
>> >>>>
>> >>
>> GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
>> >>>>
>> >>>> Leave Link 103 at Tue Mar 26 08:37:13 2019, MaxMem=10737418240 cpu:
>> >>>> 0.5
>> >>>> (Enter /scf-data/apps/gaussian/g09/l202.exe)
>> >>>> Small interatomic distances encountered:
>> >>>> 18 4 4.38D-01
>> >>>> Atoms too close.
>> >>>> Error termination via Lnk1e in /scf-data/apps/gaussian/g09/l202.exe
>> >> at Tue
>> >>>> Mar 26 08:37:14 2019.
>> >>>> Job cpu time: 0 days 0 hours 0 minutes 18.3 seconds.
>> >>>> File lengths (MBytes): RWF= 22 Int= 0 D2E= 0 Chk=
>> >> 86
>> >>>> Scr= 2
>> >>>>
>> ======================================================================
>> >>>>
>> >>>>
>> >>>> *My input file is:*
>> >>>>
>> >>>>
>> >>
>> *=========================================================================*
>> >>>> 300K constant temp QMMMMD
>> >>>> &cntrl
>> >>>> imin=0,
>> >>>> ntb=1,
>> >>>> cut=10.0,
>> >>>> ntc=1, ntf=1,
>> >>>> tempi=300.0, temp0=300.0,
>> >>>> ntt=3, gamma_ln=1.0,
>> >>>> nstlim=1000, dt=0.001,
>> >>>> ntpr=1, ntwx=1,ifqnt=1,
>> >>>> ntr=1,
>> >>>> restraintmask=':1-22', restraint_wt=5.0,
>> >>>> /
>> >>>> &qmmm
>> >>>>
>> >>>>
>> >>
>> iqmatoms=5633,5634,5635,5636,5637,5638,5639,5640,5641,5642,5643,5644,5645,5646,5647,5648,5632,5687,5688,5689,5690,5691,5692,432,433,434,435,436,437,438,439,440,441,5693,5694,5695,5696,5697,5698,5699,5700,5701,5702,5703,5704,5099,5100,5101,5102,5103,5104,5105,5106,5107,5108,5109,5110,5676,5714,5715,5716,1663,1664,1665,1666,1678,1679,1680,1681,1682,1683,1698,1699,1700,1701,1702,1703,1704,1705,1706,1707,1708,1709,1710,1721,1722,1723,1724,1725,1726,1727,
>> >>>> qmcharge=0,
>> >>>> qmshake=0,
>> >>>> qm_ewald=0, qm_pme=1,
>> >>>> qm_theory='EXTERN',
>> >>>> qmcut=10,
>> >>>> spin=1,
>> >>>> /
>> >>>> &gau
>> >>>> mem = '80GB',
>> >>>> method = 'B3LYP',
>> >>>> basis = '6-31++G(d,p)',
>> >>>> num_threads = 32,
>> >>>> use_template = 0,
>> >>>> /
>> >>>>
>> >>>>
>> >>
>> *=========================================================================*
>> >>>> _______________________________________________
>> >>>> AMBER mailing list
>> >>>> AMBER.ambermd.org
>> >>>> http://lists.ambermd.org/mailman/listinfo/amber
>> >>> _______________________________________________
>> >>> AMBER mailing list
>> >>> AMBER.ambermd.org
>> >>> http://lists.ambermd.org/mailman/listinfo/amber
>> >>>
>> >> _______________________________________________
>> >> AMBER mailing list
>> >> AMBER.ambermd.org
>> >> http://lists.ambermd.org/mailman/listinfo/amber
>> >>
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org
>> > http://lists.ambermd.org/mailman/listinfo/amber
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
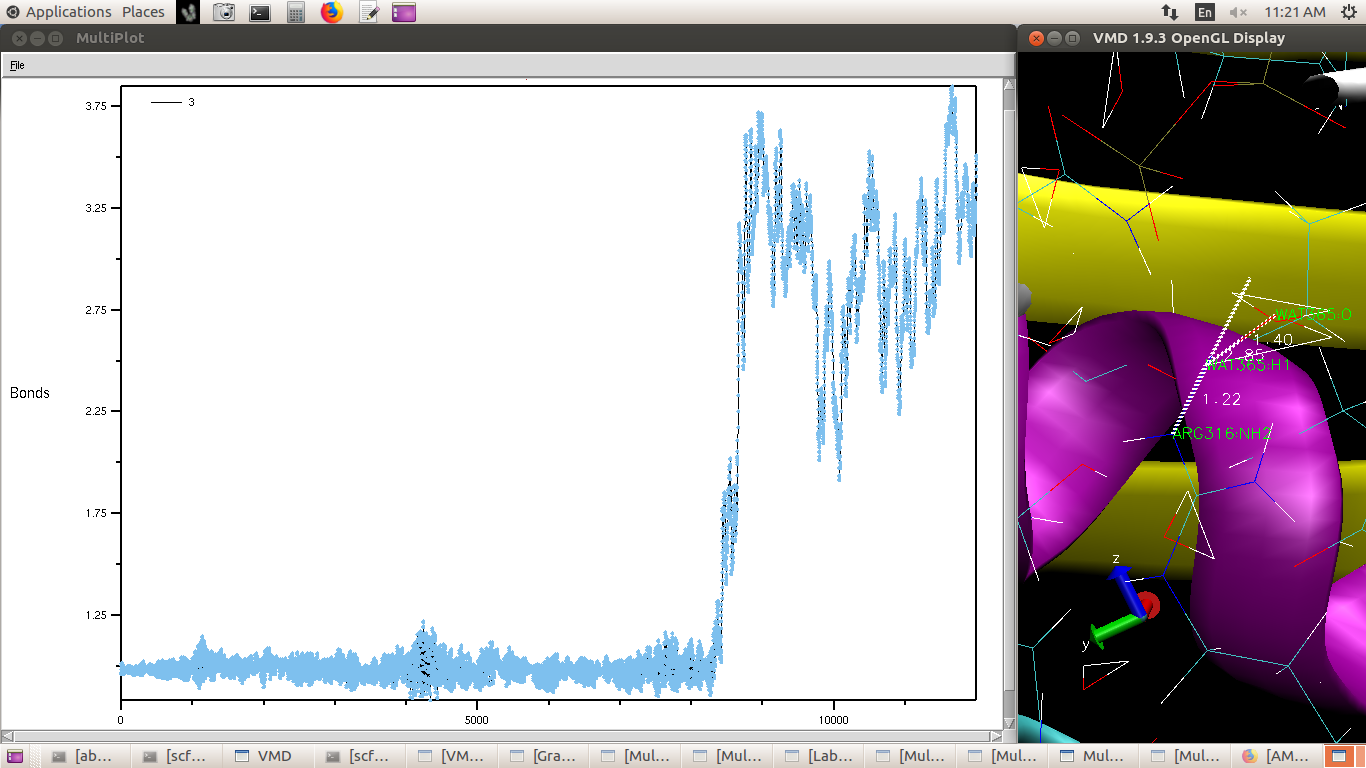
(image/png attachment: Arginine_H_bond.png)
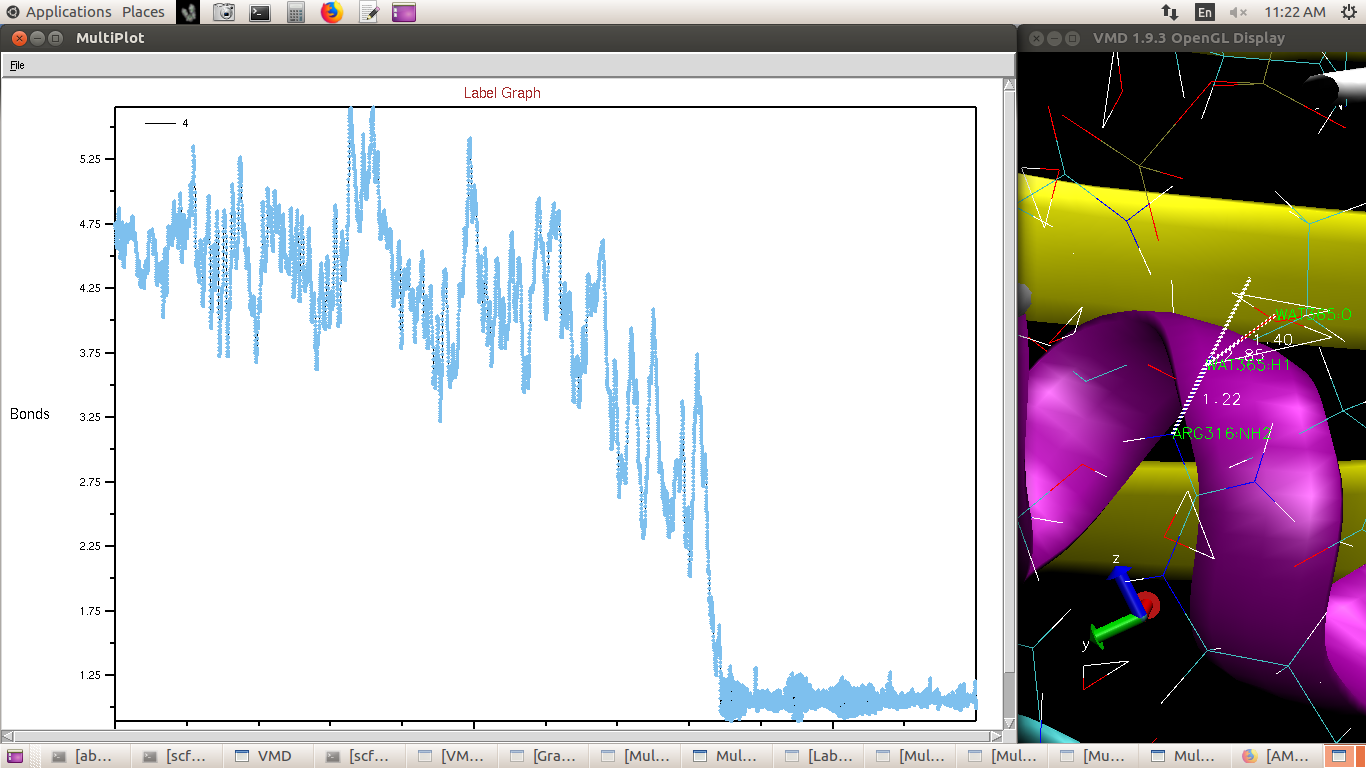
(image/png attachment: water_H_arginine_N.png)
