Date: Thu, 6 Dec 2018 20:21:19 +0000
Dear Amber Users,
I have attempted an Amber 18 PME run for an RNA fragment with multiple Mg2+ ions (X-ray data) which I
neutralized with additional magnesiums. Following Amber tutorial "Modeling a Magnesium-DNA System using
the 12-6-4 LJ-Type Nonbonded Model" (http://ambermd.org/tutorials/advanced/tutorial20/12_6_4.htm#ref3),
I built the PRMTOP and INPCRD files via LEaP and ParmEd, as follows:
1) LEaP:
source leaprc.RNA.OL3
source leaprc.water.tip3p
loadamberparams frcmod.ions234lm_1264_tip3p
mol=loadpdb RNA-7MG.pdb
check mol - net charge of -28
addIons mol MG 0 - I had to use MG; Mg2+ led to an error in LEaP
check mol
savepdb mol 2NOK_nionsMG.pdb
solvateBox mol TIP3PBOX 10.0 0.832
savepdb mol RNA7MG-solvMG.pdb
saveAmberParm mol 2RNA7MG-solvMG.prmtop RNA7MG-solvMG.inpcrd
quit
2) ParmEd:
loadRestrt RNA7MG-solvMG.inpcrd
setOverwrite True
add12_6_4 .%Mg2+ watermodel TIP3P
outparm RNA7MG-solvMG-1264.prmtop RNA7MG-solvMG-1264.inpcrd - *CCOEF section added to PRMTOP
Following the above, I started an equilibration run (repeated MIN, HEAT, MD at 300K phases with slowly released harmonic restraints,
calling pmemd.cuda and pmemd.cuda.MPI binaries). Very quickly, beginnning with the first heating for 40ps, the solvent box begins to
distort in a periodic way I have never seen before in runs with monovalent ions.
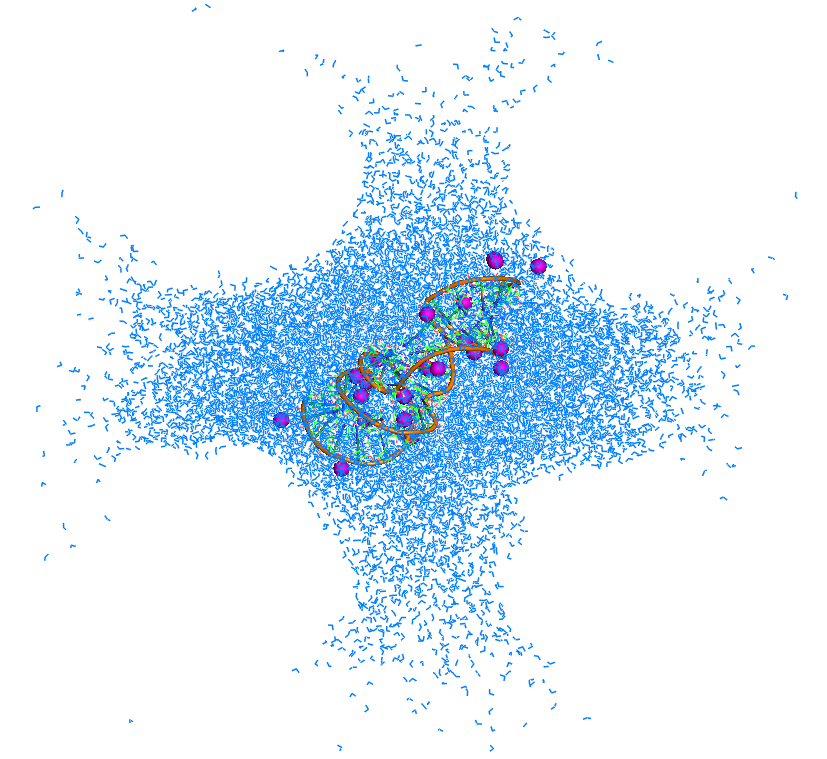
Please, refer to the attached PNG file (my earlier e-mail with 3 PDBs was too large for posting unfortunately) as the system's shape is
hard to describe. I hope that this pattern of 8 spherical voids around the original cuboid box will remind somebody of a problem they
have seen before.
While the MD terminates with errors (cudaMemcpy GpuBuffer:: Download failed an illegal memory access was encountered), probably
due to expanding size of the system, the RNA and the Mg ions stay close with three of them maintaining correct coordinating positions.
I have tested Amber 18 PME system with TIP3P waters and default Na+/Cl- ions, and it behaves correctly.
Hope to hear advice on how to fix the problem or how to debug the issue.
Best regards, Voytek Kasprzak
Wojciech (Voytek) K. Kasprzak (Contractor)
Bioinformatics Analyst
Basic Science Program
Frederick National Laboratory for Cancer Research
Leidos Biomedical Research, Inc.
Post Office Box B
Frederick, Maryland 21702
Phone: 301-846-5537
kasprzaw.mail.nih.gov
http://binkley2.ncifcrf.gov/users/kasprzak
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: PME-EQLR-0100ps-box.png)
