Date: Mon, 3 Dec 2018 11:46:28 +0530
Hello sir
As suggested by you, I studied the cpptraj cluster command in the Amber 18
manual and also followed the tutorial. I was able to perform but got two
different types of results for two different Molecular dynamics
simulations. In first case, the cluster with the maximum population is
stable and in other case the population of the dominant cluster is
decreasing with time. The DBSCAN algorithm was performed with rms
calculated between CA,C,N.
Attached are the two files.
I am confused whether both the results are correct and giving some
information or the simulations performed in the second case has to be
looked upon again. Is there a criterion to accept or reject the simulation
parameters on the bases of the stability of the dominant cluster?
Please advise and thank you for your time.
with regards
Rajbinder Kaur Virk
On Mon, 19 Nov 2018 at 7:31 PM, Daniel Roe <daniel.r.roe.gmail.com> wrote:
> Hi,
>
> Your issue is here:
>
> On Mon, Nov 19, 2018 at 7:59 AM Rajbinder Kaur Virk
> <rajbinderkaurvirk.gmail.com> wrote:
> > No clusters found.
>
> This isn't a cpptraj error per se. Based on the parameters you gave
> the 'cluster' command, no clusters were found. You'll need to try to
> adjust the parameters you're giving the DBSCAN algorithm, or try
> another algorithm. Clustering is much more of an art form than a
> science, so plan on a fair amount of trial and error here. It's good
> that you're starting off with a relatively small trajectory; this will
> allow you to become familiar with the results without waiting around
> too much. I highly recommend reading the entire 'cluster' command
> entry in the Amber 18 manual if you haven't already (section 29.11.4,
> starting on page 694), and in particular the 'Hints for setting DBSCAN
> parameters' subsection if you want to continue using DBSCAN. I also
> recommend reading this excellent paper on clustering MD trajectories
> from Shao, Cheatham et al.:
> https://pubs.acs.org/doi/abs/10.1021/ct700119m
>
> Some general clustering tips:
>
> 1) In my experience I've obtained better results with 'sieve <#>
> random' as opposed to plain 'sieve <#>'.
> 2) If you're not going to change your distance metric (including
> sieve) you way want to use 'savepairdist'/'loadpairdist' to avoid
> recalculating the pairwise distances every time. This is useful e.g.
> when doing hierarchical aggolmerative with varying epsilon, etc.
> 3) The OpenMP version of cpptraj (cpptraj.OMP) can accelerate the
> pairwise distance calculation, but don't use more threads than you
> have physical cores.
>
> Also, there is a very basic clustering tutorial you may find useful
> here: http://www.amber.utah.edu/AMBER-workshop/London-2015/Cluster/
>
> Hope this helps,
>
> -Dan
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
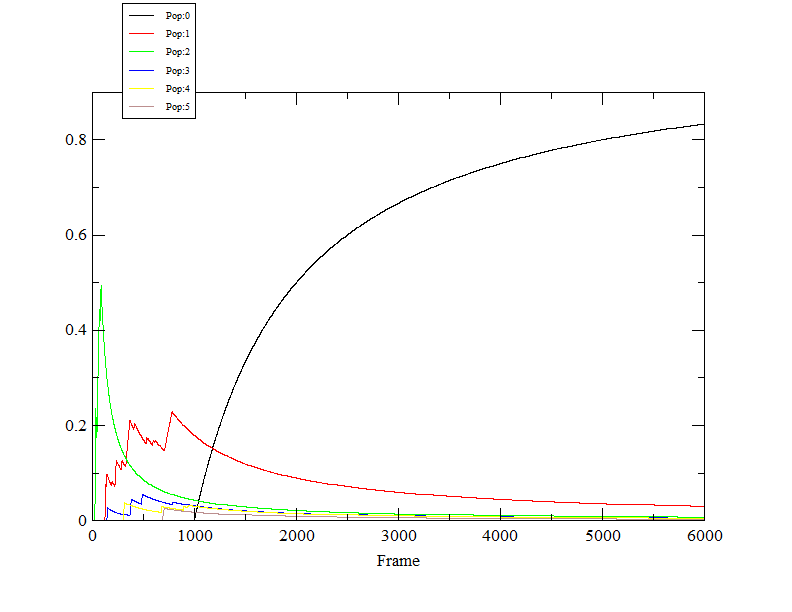
(image/png attachment: implicit_0.png)
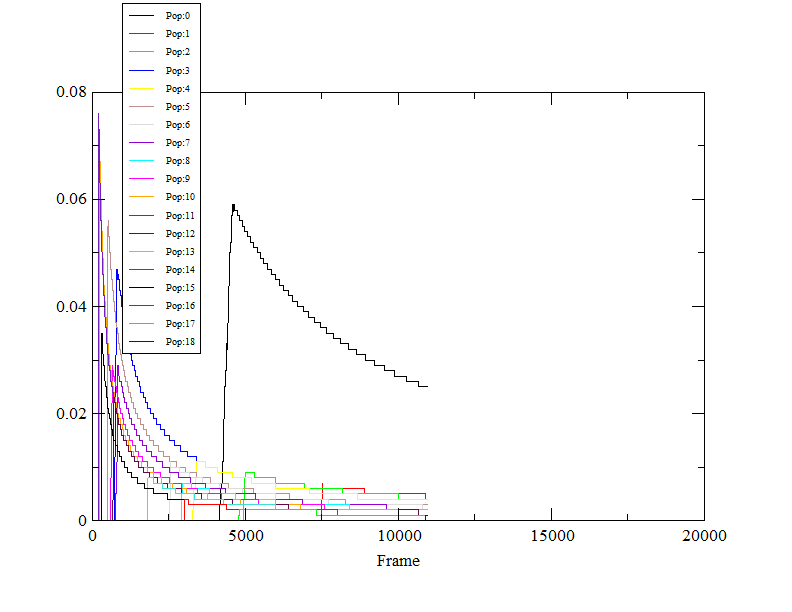
(image/png attachment: implicit_RT.png)
