Date: Mon, 11 Jun 2018 14:33:42 +0200
Dear Amber users,
After following the aMD tutorial, I carried out a 500 ns long aMD
simulation using pmemd. The unfolded peptide was solvated in water and
chloroform box.
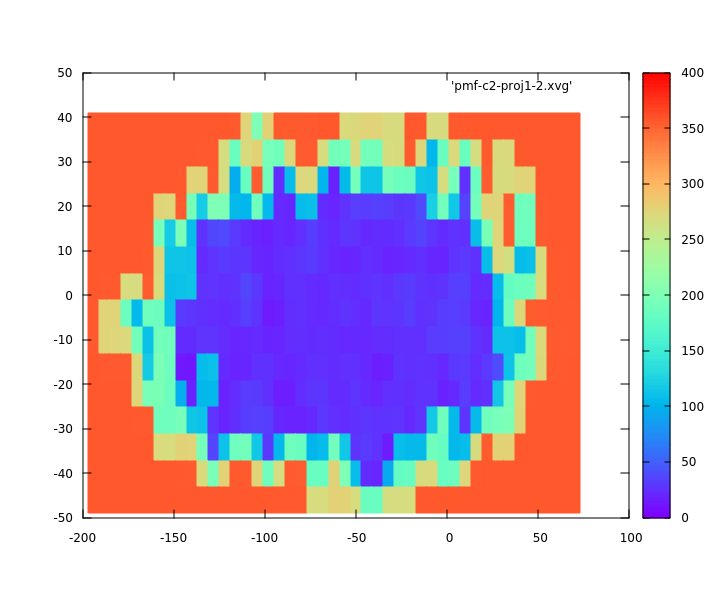
The problem is with re-weighting results. When I plot phi-psi as function
of PMF, the PMF (in kcal/mol) reaches the highest value of 350 kcal/mol in
water while remains around only 20 kcal/mol in chloroform.
Because of this, few conformations close to 0 kcal/mol exist on the free
energy surface. Till now, I could only find one more result with very high
PMF ~1200 kJ/mol which is comparable to mine. Is it normal to have such
high PMF values? What does it say about the simulation?
As I understand, by reweighting we capture the otherwise, 'unperturbed'
conformation. So, if few conformations on the free energy landscape after
removal of potential boost shows very high energy, it must mean that some
rare transition event with a really high energy barrier has occurred. Am I
missing something in this inference?
Any suggestion is well appreciated.
-- Best wishes Chetna Tyagi
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: FEL-PC1-2.png)
