Date: Sun, 3 Jun 2018 16:27:10 +0200
Here I attach the image, in case not visible with previous email...
On Sun, Jun 3, 2018 at 4:25 PM, Chetna Tyagi <cheta231.gmail.com> wrote:
> Dear all,
>
> After following the aMD tutorial, I carried out a 500 ns long aMD
> simulation using pmemd. The unfolded peptide was solvated in water and
> chloroform box.
>
> The calculation finished without any error reports and after analysis
> everything seems fine. The phi-psi plots as function of free energy also
> seem okay.
>
> The problem is with re-weighting results. When I plot phi-psi as function
> of PMF, the PMF (in kcal/mol) reaches the highest value of 400 in water
> while remains around only 20 in chloroform.
>
> Because of this, few conformations close to 0 kcal/mol exist on the free
> energy surface. Till now, I could only find one more result with very high
> PMF ~1200 kJ/mol which is comparable to mine. Is it normal to have such
> high PMF values? What does it say about the simulation?
>
> The below image is for one residue. Although, the secondary structure
> conformations are expected. What role does the solvent play in aMD ?
>
>
>
>
> Any suggestion is well appreciated.
>
> --
> Best wishes
> Chetna Tyagi,
> University of Szeged
>
>
-- Best wishes Chetna
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
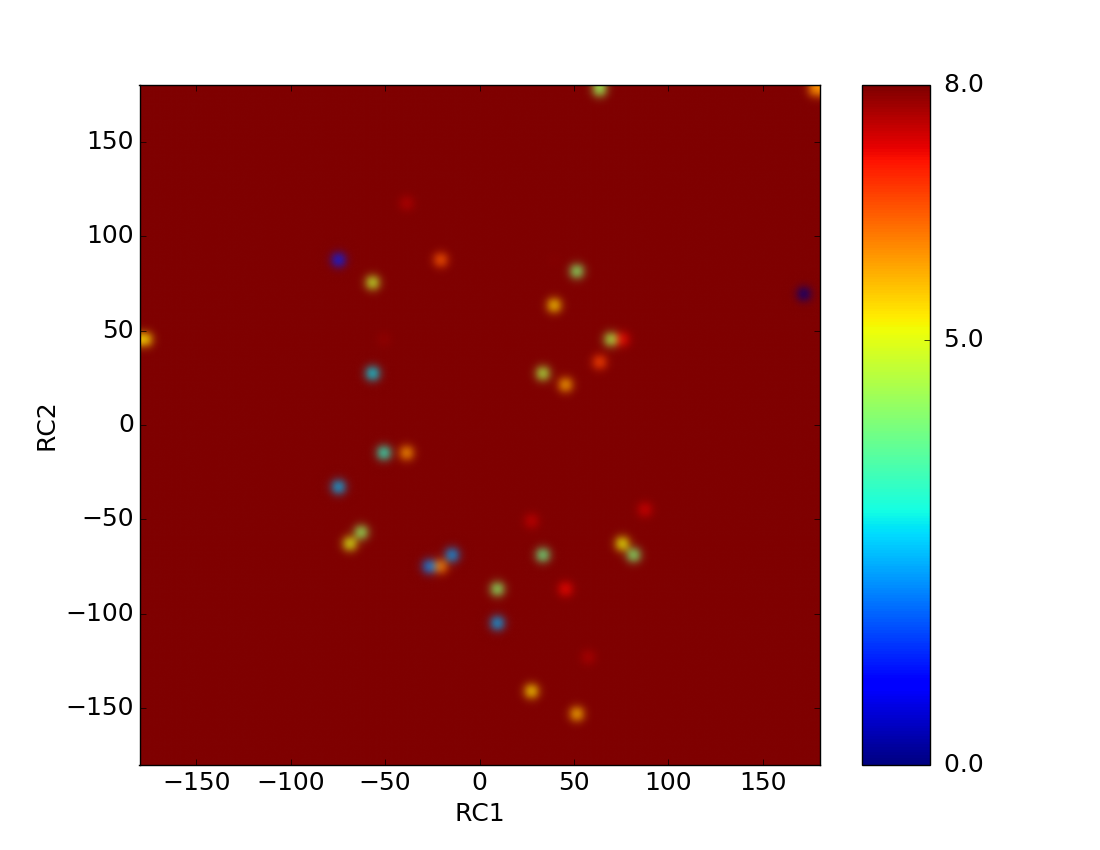
(image/png attachment: 2D_Free_energy_surface.png)
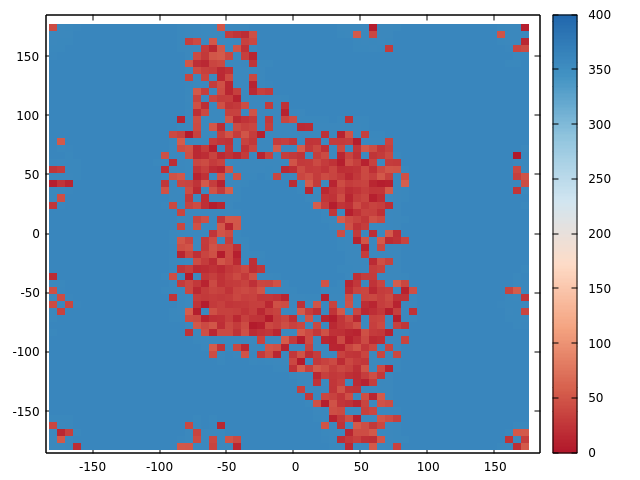
(image/png attachment: PMF-Dihed2.png)
