Date: Thu, 16 Jun 2016 12:11:39 +0800
Dear all,
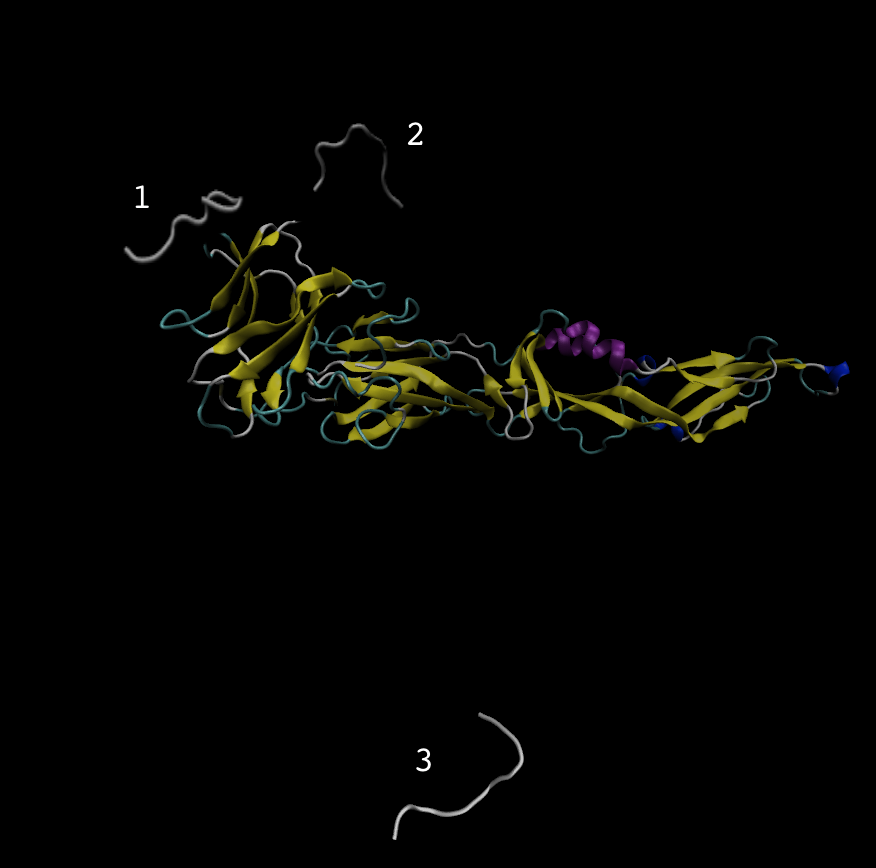
I am simulating a peptide that has known activity to a receptor, but
throughout the 20ns simulations, the peptide is no longer at the binding
site, pls see the image below, on how the peptide moved out from the
binding site to location 3.
Is this something to do with the trajectory imaging? I have tried 1.
autoimage and 2. center, image familiar, and both gave the same output.
Or the peptide just doesn't fit to the binding site? The energy at the last
.out file is still negative.
Pls advise.
Thanks!
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 3.png)
