Date: Tue, 16 Feb 2016 16:13:57 +0530
Dear all,
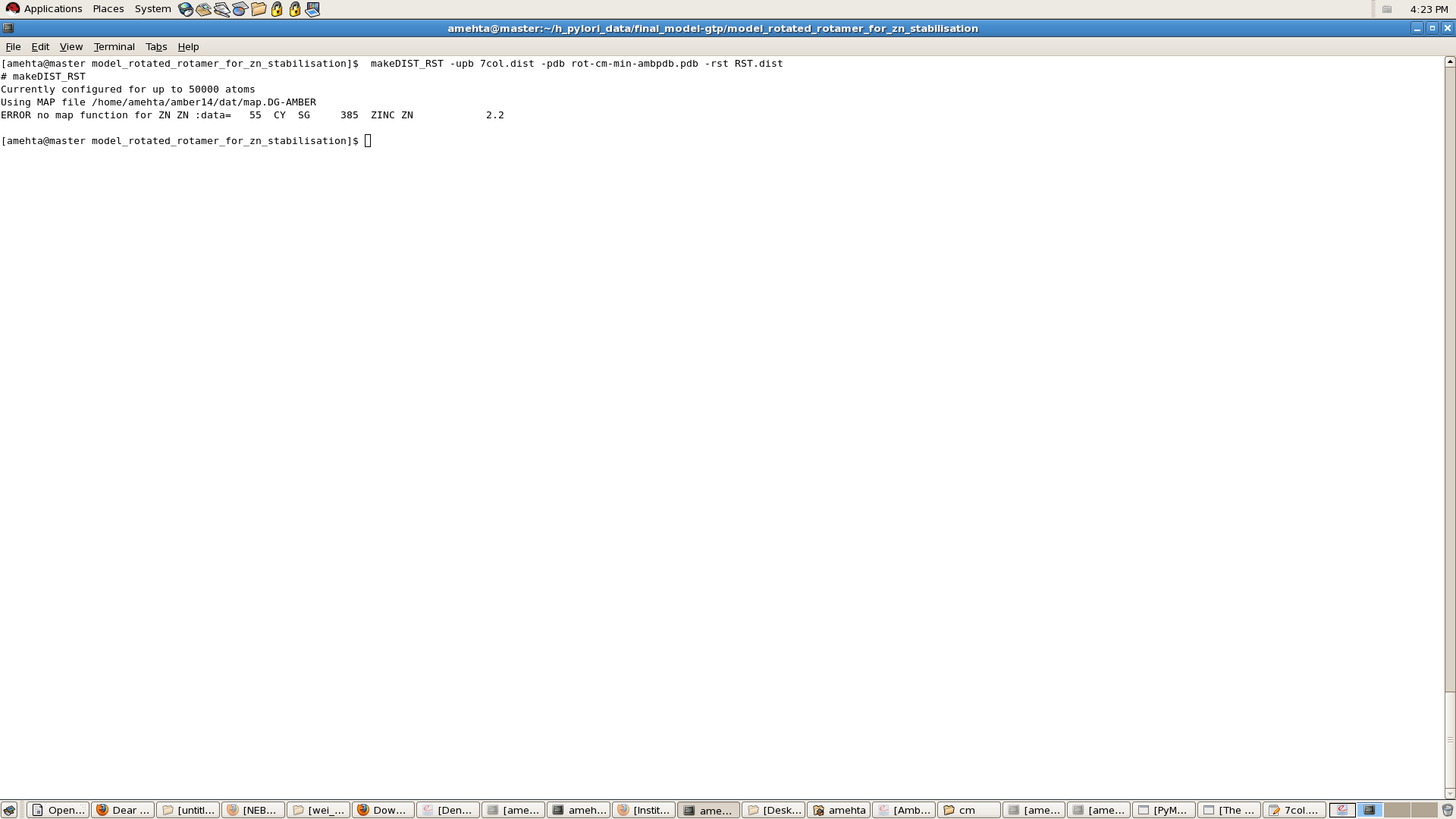
I have tried to make 7col.dist file to use it further to apply the
restraints...
But , when i run the command to conert in rst format it gives me error!
i have attached the screen shot giving the error and my 7col.dist file as
well!
Please guide!
thanks in advance
On Fri, Feb 12, 2016 at 7:51 PM, Gustavo Seabra <gustavo.seabra.gmail.com>
wrote:
> Dear Ankita,
>
> Using weak NMR restraints should work to keep the ion in place for the
> minimization…
>
> BUT,
>
> Even tough you *can* use some weak restraints to hold the Zn ion in place,
> if you plan to run other simulations on this system, I suspect the real
> question you need to address is *why* the ion is leaving the position.
>
> I suggest that you look into other simulations of metalloproteins before
> continuing. Depending on your system, there are cases where the Zn ion can
> be treated simply as the Zn2+ ion, but there also cases where you may need
> to reparameterize parts of the system to consider bonds with the Zn Ion,
> and only you can make this decision.
>
> —
> Gustavo Seabra.
>
>
>
> > Em 12 de fev de 2016, à(s) 08:03, ankita mehta <mehtaroadies.gmail.com>
> escreveu:
> >
> > Dear All,
> >
> > I want to minimize a protein which contains Zn ion.
> > But the problem is Zn ion is displacing from the position after
> > minimization!
> > Can u suggest the way through which position of Zn ion can be fixed.
> > i am attaching t he leap.log file and min.out file..
> > please suggest the way out!
> >
> > I shall be thankful!
> >
> <leap.log><min-solvation-12-6-4.out>_______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: 7col.dist
(image/png attachment: Screenshot.png)
