Date: Wed, 16 Dec 2015 08:07:55 -0600
Hi Amber-developers/users,
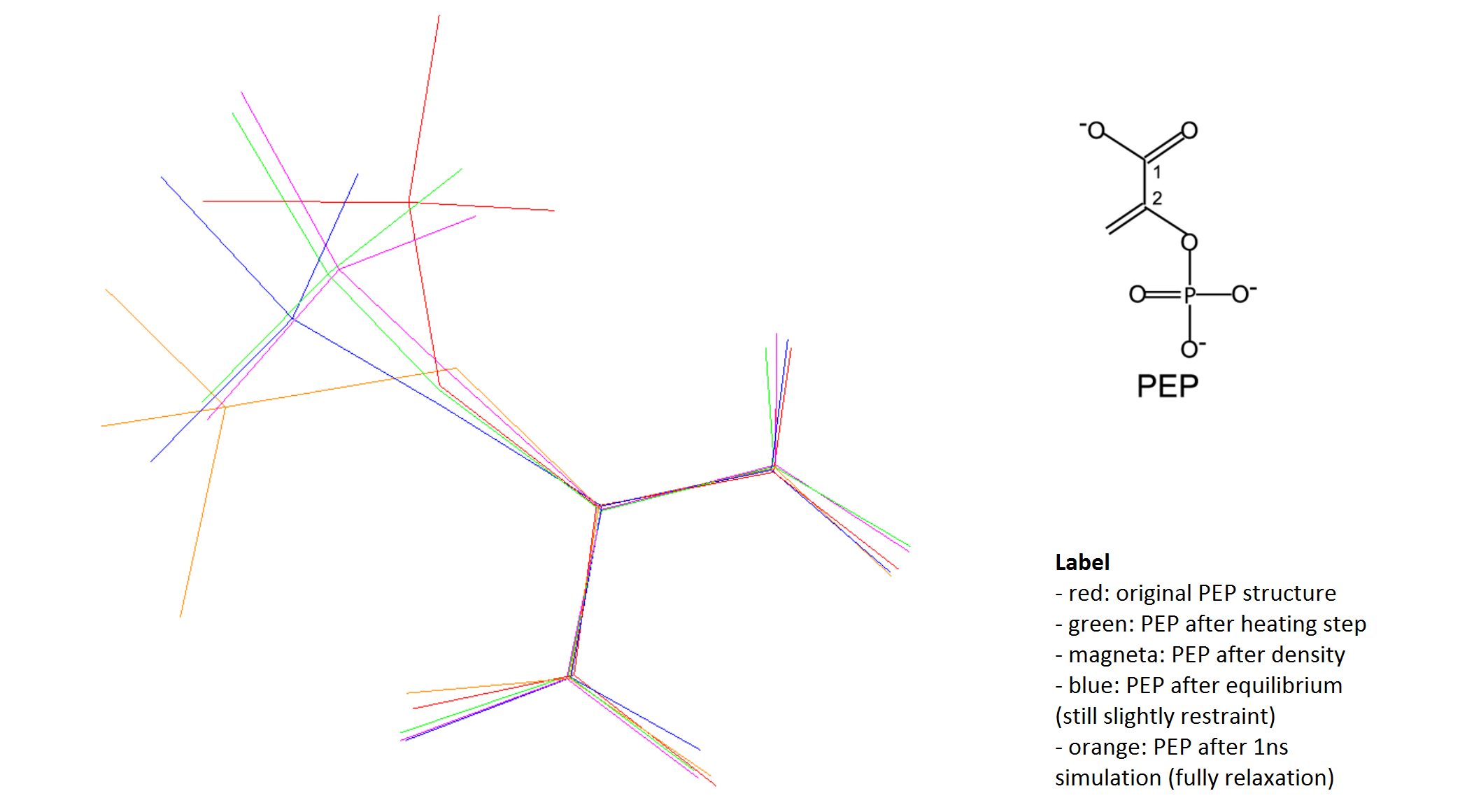
Our group currently works with Enzyme I-PTS (monomer, 313aa) and ligand PEP
(12atoms, -3 charge). The original model EI-PEP comes from our both
supposition and NMR experiments. After running MD simulation for 1ns, we
recognized that the ligand's structure has been changed significantly from
the original structure (see attached file). To our point of view, this
change in ligand's structure should not be acceptable, compared with the
experimental model. Especially in case the change is caused mainly by
solving bad contacts during system preparation. Now, we are working on
different reasons causing the problem.
- Sets of charge on ligand. We tried with different charge methods to see
which performs better during simulations. We tried to get RESP charges with
GAMESS-US output but failed. We are not sure that the providing script (of
Dr Hans De Winter) works for GAMESS-US or PC-GAMESS. We also would like to
try with R.E.D. server at http://q4md-forcefieldtools.org/REDS/ but it
requires name and password for authorization.
- Angle/bond/dihedral definition. It seems that we have to define our own
lib and frcmod files for ligand. Then the ligand's structure after 1ns
looks better (not changed so much with the experimental model). But could
you please help me to check these parameters (please see the attached files
for pdb, lib as "log" and frcmod)
- The final thing are parameters when running MD simulation from
minimization to production. We did minimize system with strong restraint on
both protein and ligand (at the first time, by following some online
tutorials, we only restraint backbone of protein), then minimize with soft
restraint and fully relax. For heating, density and equilibrium, we also
tried to perform the same thing: strong restraint then relax gradually.
Btw, can we perform restraint with different forces for different groups?
i.e. strongly restraint on ligand, and weakly restraint on backbone.
Could you please give me any suggestion. Do these approaches sound
reasonable to get through the problem.
Thank you very much
Best Regards,
Trang Nguyen
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ligands-aligment.png)
- application/octet-stream attachment: pep.frcmod
- application/octet-stream attachment: pep.log
- chemical/x-pdb attachment: pep.pdb
