Date: Thu, 29 Sep 2011 15:43:24 +0800
Dear Amber users,
I am doing unfolding for my protein, and I got the error as below:
*
forrtl: severe (64): input conversion error, unit 9, file
/gpfs/home/sutr0003/NDM1/NDM-simulation/./ndm_heat3.rst
Image PC Routine Line
Source
sander.MPI 000000000093AF9D Unknown Unknown Unknown
sander.MPI 0000000000939AA5 Unknown Unknown Unknown
sander.MPI 00000000008D6EF9 Unknown Unknown Unknown
sander.MPI 0000000000877A0D Unknown Unknown Unknown
sander.MPI 000000000087725A Unknown Unknown Unknown
sander.MPI 000000000089E96B Unknown Unknown Unknown
sander.MPI 000000000053C1AC Unknown Unknown Unknown
sander.MPI 00000000004FE6A9 Unknown Unknown Unknown
sander.MPI 00000000004DE8CA Unknown Unknown Unknown
sander.MPI 00000000004D9A17 Unknown Unknown Unknown
sander.MPI 000000000042F3AC Unknown Unknown Unknown
libc.so.6 000000392501D974 Unknown Unknown Unknown
sander.MPI 000000000042F2B9 Unknown Unknown Unknown
--------------------------------------------------------------------------
mpirun has exited due to process rank 0 with PID 25993 on
node compute169 exiting without calling "finalize". This may
have caused other processes in the application to be
terminated by signals sent by mpirun (as reported here).
--------------------------------------------------------------------------
Unit 30 Error on OPEN:
./ndm_heat4.rst
--------------------------------------------------------------------------
MPI_ABORT was invoked on rank 0 in communicator MPI_COMM_WORLD
with errorcode 1.
NOTE: invoking MPI_ABORT causes Open MPI to kill all MPI processes.
You may or may not see output from other processes, depending on
exactly when Open MPI kills them.
--------------------------------------------------------------------------
--------------------------------------------------------------------------
mpirun has exited due to process rank 0 with PID 26018 on
node compute169 exiting without calling "finalize". This may
have caused other processes in the application to be
terminated by signals sent by mpirun (as reported here).
--------------------------------------------------------------------------*
After refered to some previous threads which explained that the errror might
be involved in testing process during installation, but I don't think it is
my case because:
- The testing process was done
- I can read/write in my directory
Another suggestion could be the solvent box. It might be not sufficient when
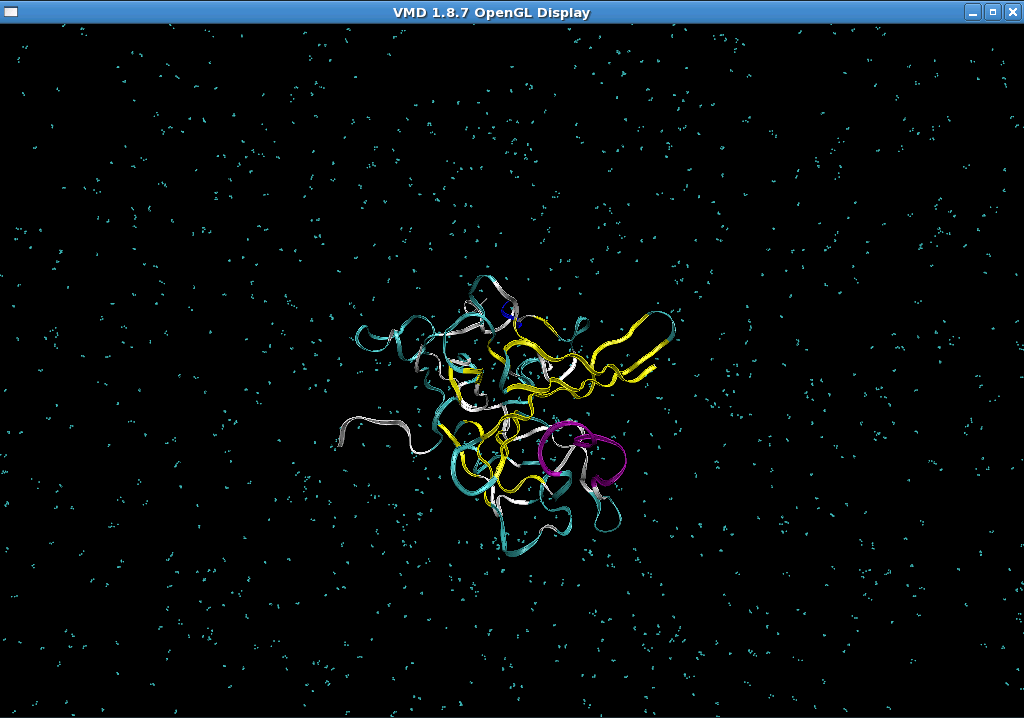
the molecule is extended. However, as I visualized the mdcrd file during
heating, my molecule is still located inside the box (the attached image).
Could anyone please help give me some suggestion? I wanted to unfold my
protein ( in 60ns, but got the error at the 18th ns.
Thank you for any help.
Regards,
Chinsu
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: heat3.png)
