Hi all,
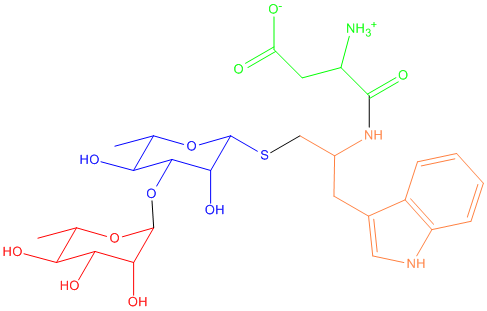
I am trying do set parameters for a ligand as shown in the attachment with
GLYCAM, GAFF, and 99SB force fields for different parts.
For the left part (one standard alpha-L-Rhamnose and a thio
alpha-L-Rhamnose), I calculated RESP charge and used antechamber to set GAFF
parameters for the modified thio sugar (blue part). Then I loaded GLYCAM_06
and GAFF ff within tleap, after which I loaded the standard sugar (red part)
and the modified thio sugar (blue part).
But after I linked these two together, missing parameters remained for the
newly-formed glycosidic bond and angles and dihedrals, as expected. So I
wonder, technically, how to fill in these missing parameters. Should I
manually construct a .frcmod file? Or is there a better alternative to fuse
these different parts?
Thanks for any suggestion.
Yun
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Aug 26 2011 - 18:30:03 PDT
{kind=link}
