Sorry that I should have attached this model molecule I want to study
earlier.
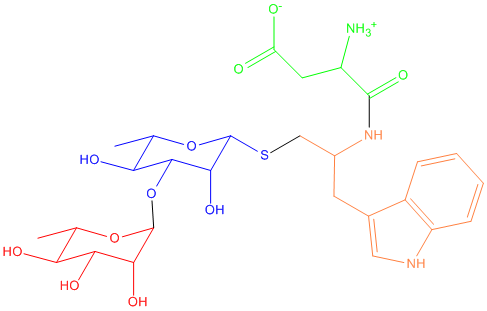
As you can see, I divide this ligand into four parts, and the parameters
(atomic charges, bonds, angles, dihedrals, impropers) of the red (terminal)
sugar and green ASP can be obtained from GLYCAM and 99SB respectively.
But I have to include the glycosidic oxygen between two sugar rings in the
blue thio sugar, as this glysicidic oxygen is defined in the preceding sugar
(the blue thio sugar) instead of the succeeding sugar according to GLYCAM.
I plan to use the approach -2- you mentioned (so that I don't need generate
many conformations), but considering my specific case, I wonder why not
using
Me-O3-L-RhamnoS-Me + MeS-CH2CH(R)NH-COMe
<----------------------> <===========>
INTER-MCC=0 INTRA-MCC=0
, and then remove the Me - Me and MeS - COMe respectively?
Since the L-Rha and TRP in my case carry only one modified site compared to
their standard counterparts, should I include charge constraints (keep the
atomic charges) for atoms far away from the modified sites? Such as C5 and
C6 in the thio sugar, and the entire indole ring of the modified TRP?
Thank you so much!
Yun
On Thu, Aug 25, 2011 at 11:37 PM, FyD <fyd.q4md-forcefieldtools.org> wrote:
> Yun,
>
> > Sorry, I just read the tutorial, that is, "Central fragment of a xxx".
> >
> > So for the modified (C-terminal reduced) amino acid, should I add a MeCO
> at
> > the N-terminus and MeS at the reduced C-terminus? (considering the
> reduced
> > C-terminus is used to link glycosidic sulfur atom).
> >
> > And for the sugar molecule, should I add a Me cap to 3-hydroxyl group in
> > addition to a methyl group attached to the glycosidic sulfur? (This sugar
> > molecule would fall into the category of central fragment of my ligand)
>
> ups I overlooked your problem ;-) You do not have any cysteine residue...
>
> Let's re-do the explanations with your terminal molecule/pseudo amino-acid:
>
> -1- you consider the following molecule
>
> Thio-L-Rhamno-CH2CH(R)NH-COMe ; R = CH2-Ph
> <==>
> INTRA-MCC
>
> You only have to set up an intra-molecular charge constraint
> (INTRA-MCC keyword) for the COMe capping group (see <==> above).
> See http://q4md-forcefieldtools.org/Tutorial/Tutorial-3.php#15
>
> R.E.D. will generate a sm fragment (mol2 file) for you.
>
> -2- you split your molecule into two building blocks.
>
> L-RhamnoS-Me + HS-CH2CH(R)NH-COMe
> <-------> <==>
> INTER-MCC INTRA-MCC
>
> See http://q4md-forcefieldtools.org/Tutorial/Tutorial-3.php#16
> or http://q4md-forcefieldtools.org/Tutorial/Tutorial-3.php#17
>
> You set up an intra-molecular charge constraint (INTRA-MCC keyword)
> for the COMe capping group.
> You set up an inter-molecular charge constraint (INTER-MCC keyword)
> between the methyl group of L-RhamnoS-Me and the thiol group of your
> pseudo terminal amino-acid.
>
> The approach -2- is once again more complex but more flexible...
>
> R.E.D. will generate a FG fragment (mol2 file) in the Mol_MM directory.
>
> Sorry for misunderstanding your problem in my first email.
>
> regards, Francois
>
>
> > On Thu, Aug 25, 2011 at 9:58 AM, Yun Shi <yunshi09.gmail.com> wrote:
> >
> >> Thank you very much!
> >>
> >> I looked at the CD project, and I saw when FFTopDB were constructed, the
> >> 1-metylated glucose was used to derive RESP charges. So in my
> >> thio-glycopeptide case, should I use the sugar molecule with a methyl
> group
> >> attached to the glycosidic sulfur as well?
> >>
> >> What about the modified (C-terminal reduced) amino acid? Add one more
> >> methyl group to the modified C-terminal and acetate to the N-terminal?
> >>
> >> But in the end, what is deposited in the FFTopDB is the residue without
> the
> >> methyl or acetate cap, right?
> >>
> >> Yun
> >>
> >>
> >>
> >> On Wed, Aug 24, 2011 at 11:38 PM, FyD <fyd.q4md-forcefieldtools.org
> >wrote:
> >>
> >>> Dear Yun Shi,
> >>>
> >>> > I am trying to understand how this works.
> >>>
> >>> If you look at the data available in the "F-85" R.E.DD.B. project, you
> >>> will find a x/tLEaP script to construct the CD-based glycopeptides as
> >>> well as a frcmod file for missing force field parameters with comments.
> >>> http://q4md-forcefieldtools.org/REDDB/projects/F-85/script1.ff
> >>> http://q4md-forcefieldtools.org/REDDB/projects/F-85/script3.ff
> >>>
> >>> > So instead of combining individual residues in a building-block
> manner,
> >>> as
> >>> > in the assignment of atomic charges for proteins with amber99sb, it
> is
> >>> > recommended to consider the ligand as a holistic molecule when
> >>> calculating
> >>> > the RESP charge?
> >>>
> >>> 'recommended'? ;-) ... Personally, I use most of the time the building
> >>> block approach whatever if the target 'big' molecule is a ligand or a
> >>> nucleic acid/protein/polysacharide.
> >>>
> >>> > I am curious that if I could do things in a building-block
> >>> > manner since it can potentially decrease a lot of computational time
> for
> >>> > geometry optimization.
> >>>
> >>> The building-block approach has many advantages:
> >>> - it potentially "decreases a lot of computational time for geometry
> >>> optimization" as you said.
> >>> - it allows rigorously defining the conformation of each
> >>> building-block and not to use a conformation more or less randomly
> >>> chosen.
> >>> - it allows avoiding interactions between charges group during
> >>> geometry optimization in gas phase.
> >>> - it allows the construction of analogs for the target molecule.
> >>> - it allows the construction of oligomers/polymers for the target
> >>> molecule.
> >>>
> >>> However, it also has disadvantages:
> >>> - it is complex to set up when one starts, but R.E.D. has been
> >>> designed for this approach.
> >>> - errors during the charge fitting step are introduced when using the
> >>> building-block approach; these errors have to be minimized by
> >>> correctly selecting the connecting groups between the different
> >>> building-blocks. The statistics module available in R.E.D.
> >>> Server/R.E.D. IV also helps to localize/minimize these errors.
> >>>
> >>> > And when it comes to geometrical parameters, we should use GLYCAM for
> >>> sugar
> >>> > part, 99SB for standard amino acids, and GAFF for organic part?
> >>>
> >>> Yes
> >>>
> >>> - We only select 'obvious' missing force field parameters from GAFF
> >>> (we recompute key dihedrals), and when used we always rationalize
> >>> these force field parameters as it was done in the Cornell at al.
> >>> force field.
> >>>
> >>> - In this work, we used Amber scaling factor values for 1-4
> >>> non-bonding interactions for all the glycopeptide molecular systems;
> >>> i.e. we did not split the system into a peptide and a sugar parts.
> >>>
> >>> > BTW, could you tell me how to generate multiple conformations with
> >>> geometry
> >>> > optimization from Gaussian 09?
> >>>
> >>> You could do a conformational search - although if the building-block
> >>> approach is used the conformational search is quite simplified...
> >>> We also often modify a key dihedral to look for lowest minimum/minima.
> >>>
> >>> To create a P2N file with multiple conformations, see:
> >>> http://q4md-forcefieldtools.org/Tutorial/Tutorial-1.php#3
> >>>
> http://q4md-forcefieldtools.org/Tutorial/Tutorial-1.php#EXAMPLE-P2N-FILE
> >>>
> >>> To create a QM file with multiple conformations to be used in the Mode
> >>> 2 of R.E.D. (see
> >>> http://q4md-forcefieldtools.org/REDS/popup/popredmodes.php), simply
> >>> concatenate the different QM outputs into a single file.
> >>>
> >>> regards, Francois
> >>>
> >>> >> Dear Yun,
> >>> >>
> >>> >> > Is it technically possible to do it due diligence in the first
> place?
> >>> >> That
> >>> >> > is, cut the molecule into three parts as I mentioned before, use
> >>> GLYCAM
> >>> >> for
> >>> >> > the sugar part, 99SB for the Thr, and GAFF for modified Phe and
> the
> >>> >> > thio-glycosidic linkage. And may I then link these parts together
> >>> using
> >>> >> LEaP
> >>> >> > ?
> >>> >>
> >>> >> Concerning the use of GLYCAM + GAFF + Amber99SB you might be
> >>> >> interested by looking at the following paper:
> >>> >> http://www.ncbi.nlm.nih.gov/pubmed/21792425
> >>> >> & its corresponding R.E.DD.B. project .
> >>> >> http://q4md-forcefieldtools.org/REDDB/projects/F-85/ + its LEaP
> >>> script:
> >>> >> http://q4md-forcefieldtools.org/REDDB/projects/F-85/script1.ff
> >>> >>
> >>> >> This work is about cyclodextrin based-glycopeptide and 1-4
> non-bonding
> >>> >> interactions in GLYCAM & Amber99SB.
> >>> >>
> >>> >> Your structure is not a cyclodextrin but this work describe (i) how
> to
> >>> >> derive charges and build force field libraries for new fragments by
> >>> >> using R.E.D. IV and (ii) proposes new directions concerning the
> >>> >> treatment of 1-4 non-bonding interactions in the context of
> >>> >> glycopeptides.
> >>> >>
> >>> >> Finally, in the LEaP script you will find examples how to connect
> >>> >> organic, amino-acid and monosaccharide units...
> >>> >>
> >>> >> regards, Francois
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Aug 26 2011 - 11:30:12 PDT
{kind=link}
