Date: Fri, 20 Aug 2010 00:55:21 -0700 (PDT)
Referring back to a problem described by:
Balvinder Singh (Wed Sep 08 2004 - 15:49:04 PDT)
I have rediscovered this problem in minimizations of 30 complexes
of 2 proteins that I minimized with the following protocol: initial
minimiation with strong restraints in vacuo, then with restraints
using GB, then unrestrained with GB. The problem shows up in the GB
minimizations, mostly in the final, long, unconstrained one. Unlike the
industrious Balvinder Singh, I only printed
out the energies every 10 steps. My goal was to have all 30
complexes minimized to the same point (as measured by drms), to see if I
could distinguish 'good' from 'bad' complexes by comparing their energies
(all complexes are of the same 2 molecules, 2 proteins made up of standard
amino acids with no other cofactors).
I used Amber 10, Forcefield was leaprc.ff94
For the final unrestrained minimization, I used
GB Energy Minimization
&cntrl
imin = 1, maxcyc = 10000,
ntmin = 1, ncyc = 20,
ntpr = 10,
ntb = 0,
igb = 1,
cut = 999.9
drms = 0.00001
The drms is rather stringent, so none of the minimizations got there, but,
looking at my minimization, a criterion of
drms = 0.01 or even 0.001 is not good enuf,
the energy could still drop a few kcal/mol further.
My problem is that the bond (and to a lesser extent valence) energy spikes
at steps xx30 and xx80. Its likely that, as Balvinder Singh found,
they also occur at steps xx05 and xx55, i.e. every 25 steps, but I did
print out the energies at those steps. The spikes do not occur all the time,
in fact they don't appear in the first 2 thousand steps or so. But then, as the
minimization goes on, both the size and frequency of the spikes increases. I
have a plot of the energies for all 30 complexes (attached) that shows the
problem gets steadily worse as the minimization proceeds. My bond
energy after about step 1000 was generally about 168, except for
steps xx30 and xx80, where it would go up a small amount (say 2-4 kcal)
or a very large amount (50 to more than 600 kcal). For example:
NSTEP ENERGY RMS GMAX NAME NUMBER
9420 -1.1843E+04 9.2814E-03 2.8792E-01 CD 1625
BOND = 168.3836 ANGLE = 739.8685 DIHED = 1302.8428
VDWAALS = -2835.5918 EEL = -23010.7186 EGB = -3978.4321
1-4 VDW = 940.2447 1-4 EEL = 14830.0193 RESTRAINT = 0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
9430 -1.1056E+04 1.8257E+01 6.1400E+02 CD 1625
BOND = 797.9902 ANGLE = 889.1781 DIHED = 1306.3816
VDWAALS = -2836.4958 EEL = -23002.0935 EGB = -3975.0551
1-4 VDW = 940.5651 1-4 EEL = 14823.1138 RESTRAINT = 0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
9440 -1.1843E+04 1.6190E-02 3.0229E-01 CD2 2609
BOND = 168.4287 ANGLE = 739.9444 DIHED = 1302.8351
VDWAALS = -2835.6302 EEL = -23010.5702 EGB = -3978.4084
1-4 VDW = 940.1704 1-4 EEL = 14829.8270 RESTRAINT = 0.0000
So you get this huge hit to the RMS (9.2814E-03 at step 9420 going to
1.8257E+01 at step 9430) that only gets partially repaired at step
9440. Seems like this must be making the minimization much less efficient.
After 10000 steps of a 4800 atom system (all standard protein residues) I am
at
NSTEP ENERGY RMS GMAX NAME NUMBER
10000 -1.1844E+04 2.2508E-02 6.7566E-01 CD 4525
BOND = 168.4205 ANGLE = 739.7665 DIHED = 1302.4882
VDWAALS = -2835.9819 EEL = -23010.4237 EGB = -3978.8126
1-4 VDW = 940.0621 1-4 EEL = 14830.2729 RESTRAINT = 0.0000
(and yes, step 9980 had a big spike in bond and valence energy)
The responses that Balvinder Singh got were not very satisfying.
Ross Walker replied that something was probably wrong with the system
or the installation of Amber. I agree that something is wrong, but
I dont think there is a problem with either the installation or
my particular system.
Instead i am wondering if its a problem that is only seen if you
1) have ntpr set so that you look at energies of steps xx05, xx30,
xx55, or xx80 AND
2) do a sufficiently long (conjugent gradient) minimization.
I have not tried the AMBER test cases, but I have been running AMBER
a long time, previously versions in the Case lab. Unfortunately,
all of my old outputs had ntpr=100 or greater, so if the problem
occurred i would not see it.
Could it be the system? Well, i know that my protein-protein
complexes had some close contacts, though they relaxed well during
the initial restrained minimizations. Just in case, i took a look at the
atoms with the GMAX for the bad steps, thinking they might be
in the interface.
I looked at steps ending in 30 and 80 that showed no jumps after
the bond energy had hit 167. The GMAX atom for these steps
varied a lot in type - H, N, C - residue, etc.
I looked at about 50 steps numbered xx30 and xx80 that showed funny spikes,
from little (2-4) to big jumps, the corresponding GMAX atoms ALL were either
ASP CG or GLU CD
So the same atom type. Most were NOT in the interface
and they could be from either molecule.
I ran my vacuum minimization for just 200 steps, and did not see the
spike problem there, but Balvinder Singh had the problem with both
igb = 1 and using a water box, I believe, so it may not be GB
dependent.
The other response was from Andreas Svrcek-Seiler
''Sometimes the minimization routine does a trial step, which
might be "too large", resulting in energy spikes. Whether (and how often)
that happens depends on the algorithm as well as on implementation
details, however it is normal and harmless.
regards
Andreas''
Since this problem happened for both me and Balvinder Singh, both using
a very typical and straightforward protocol, with conjugent gradient
minimization, i am not convinced. Especially since the problem becomes
worse as the system becomes more minimized. The advantage of GB is that
it takes solvation into account, so comparing relative energies of
my complexes might be useful. However, knowing that my complexes had
large and varying jumps in energy at step 9980 makes the whole thing
seem less convincing. Its too bad, because the energy comparison does
appear to favor correct (RMSD < 4A) over incorrect (RMSD > 20A) complexes.
I am happy to send coords and parameter files, but it would be nice if
someone tried a familiar ~5000 atom system, 10000 steps of standard
minimization, with GB on, printing out energies every 5 steps (to catch the
xx05 and xx55 spikes too). If the spikes show up there, then start to
worry about if its more general problem. If the problem does not show up,
then maybe I can get some help figuring out why my system has the
problem.
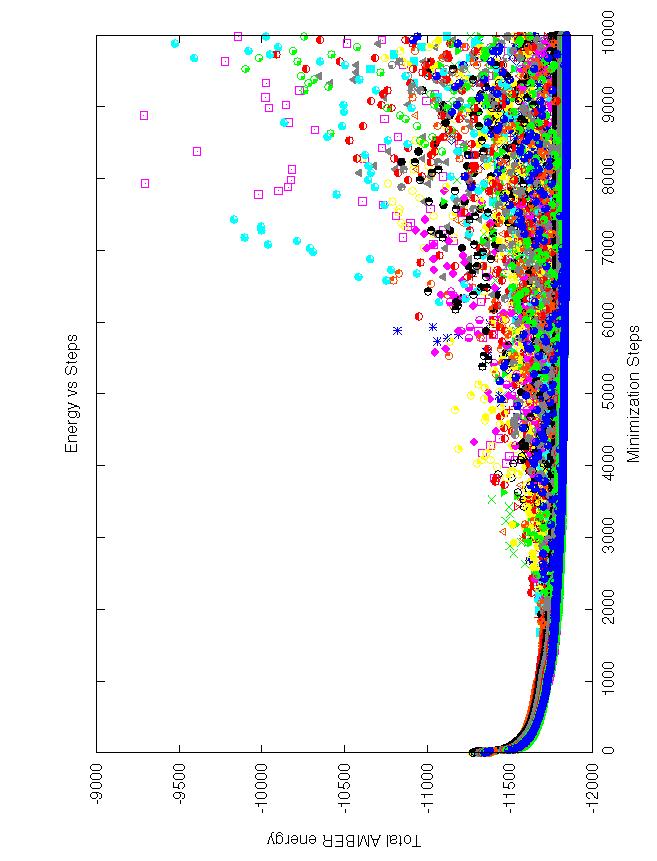
Attached is a plot of the total energy for all 30 minimizations saved
every 10 steps. It makes it very clear that the problem gets worse
as the minimizations proceed. All complexes have final minimized energies
between -11,700 and -11,900. ALL of the energies that are not close to
the minimized energy occur at steps xx30 and xx80.
Thanks,
vickie
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(IMAGE/jpeg attachment: energy.all.jpg)
