Date: Tue, 16 Dec 2025 11:01:05 -0600
Hi all,
I am looking for some advice on umbrella sampling (US) and WHAM
calculations.
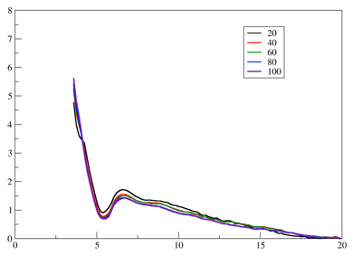
I am computing PMF along an intermolecular distance between an amino-acid
side chain and a small molecule. The US windows in the histogram show good
sampling and nice overlap, but the WHAM PMF consistently approaches ~0 at
the maximum distances (see the figure). For example, if I rerun WHAM using
only the subset of windows covering 2–10 Å, the PMF becomes 0 at d =10
instead of ~0.75 kcal/mol that we see when lotting up to d=20.
I have a large enough waterBox and PBC. I am not using periodic coordinates
in WHAM either.
Any guidance on what to check would be appreciated.
Best,
Mish
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
