Date: Thu, 13 Oct 2022 21:20:35 +0800
Dear Amber developers and users,
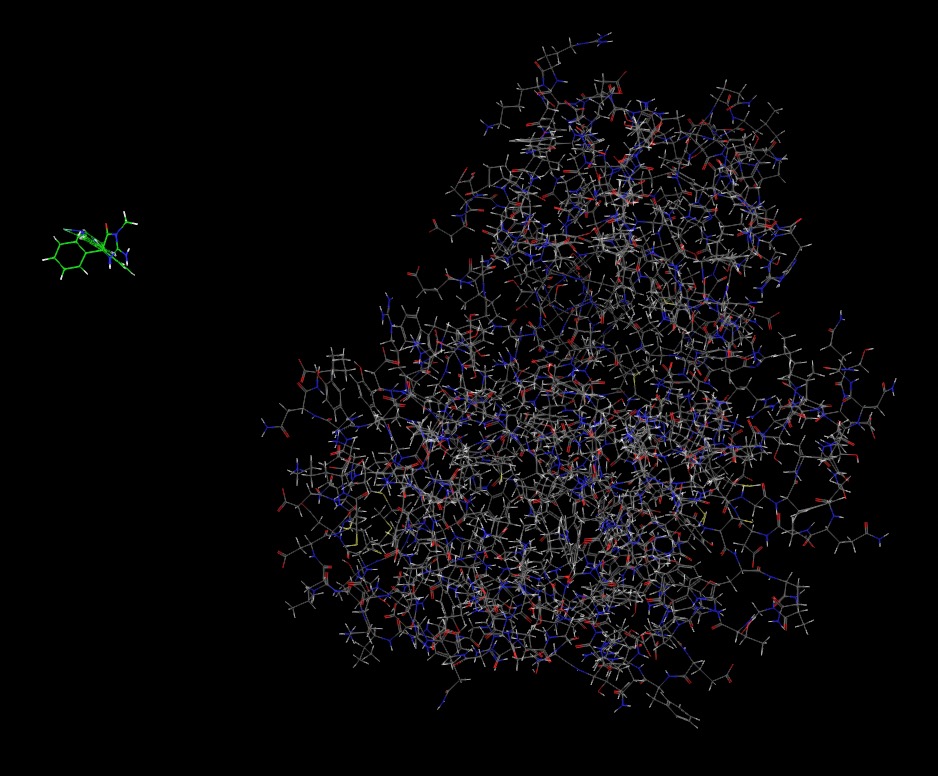
I encountered a separation of protein and ligand during TI simulation.
I chose 12 windows for lambda. At the 6th window, the ligand is separated
from the protein and almost out of the water box. It may not be related to
the periodicity because I tried to drag it back with autoimage but failed.
I affixed the picture of the structure with lambda as 0.43738 here, which
should have the ligand at the center of the protein (water box is hiden). I
also affixed my input files below the picture. I was wondering if you could
give me any suggestions on this. I really appreciate your help!
[image: image.png]
*The following are my MD input files and tleap input file.*
*minimization*
&cntrl
imin = 1, ntmin = 2, maxcyc = 1000,
ntpr = 100, ntwe = 100,
dx0 = 1.0D-7,
ntb = 1,cut=10,
icfe = 1, ifsc = 1, logdvdl = 0,
timask1=':1',
timask2=':2',
scmask1=':1.C15,C16,C17,C18,C19,N4,H14,H15,H16,H17',
scmask2=':2.C15,C16,C17,C18,C19,N4,H14,H15,H16,F1',
clambda=0.43738,
&end
/
*Heating*
&cntrl
imin = 0, nstlim = 10000, irest = 0, ntx = 1, dt = 0.002,
ntt = 1, temp0 = 300.0, tempi = 0.0, tautp = 1.0,
ntc = 2, ntf = 1,
ntb = 1, cut=10,
iwrap=1, ioutfm = 1, iwrap = 1,
ntwe = 1000, ntwx = 1000, ntpr = 1000, ntwr = 1000,
restraint_wt = 5.0,
restraintmask = '!:WAT & !.H= ',
icfe = 1, ifsc = 1, logdvdl = 0,
timask1=':1',
timask2=':2',
scmask1=':1.C15,C16,C17,C18,C19,N4,H14,H15,H16,H17',
scmask2=':2.C15,C16,C17,C18,C19,N4,H14,H15,H16,F1',
clambda=0.43738,
&end
/
&wt type='TEMP0', istep1=0, istep2=8000,
value1=0.0, value2=300.0/
&wt type='END'/
&ewald
/
*Equilibriation*
&cntrl
imin = 0, nstlim = 10000, irest = 1, ntx = 5, dt = 0.002,
ntt = 3, temp0 = 300.0, gamma_ln = 2.0, ig = -1,
ntc = 2, ntf = 1,
ntb = 2, cut = 10, ntr = 0,
ntp = 1, pres0 = 1.0, taup = 2.0,
ioutfm = 1, ntxo = 2, iwrap = 0,
ntwx = 2000, ntpr = 500, ntwr = 2000,
iwrap=1, ioutfm=1, ntwv=-1,ntave=1000,
icfe = 1, ifsc = 1,logdvdl = 0,
timask1=':1',
timask2=':2',
scmask1=':1.C15,C16,C17,C18,C19,N4,H14,H15,H16,H17',
scmask2=':2.C15,C16,C17,C18,C19,N4,H14,H15,H16,F1',
clambda=0.43738,
&end
/
*Production*
&cntrl
imin = 0, ntx = 5, irest = 1,
ntpr = 1000, ntwx = 10000, ntwe = 1000, ntwr = 1000, ig = -1,
ntp = 1, pres0 = 1.0, ntf = 1, ntb = 2, cut = 10.0,
iwrap = 1, nsnb = 10,
nstlim = 500000, t = 0.0, nscm = 1000, dt = 0.002,
temp0 = 300.0, tempi = 300.0, ntt = 3,
gamma_ln = 2.0, tautp = 2.0,
ntc = 2, ioutfm=1, ntwv = -1, ntave = 1000,
icfe = 1, ifsc = 1,ntxo = 2,
scalpha = 0.5, scbeta = 12.0, logdvdl = 0,
ifmbar = 1, mbar_states = 12, mbar_lambda = 0.00922, 0.04794, 0.11505,
0.20634, 0.31608, 0.43738, 0.56262, 0.68392, 0.79366, 0.88495, 0.95206,
0.99078,
timask1=':1',
timask2=':2',
scmask1=':1.C15,C16,C17,C18,C19,N4,H14,H15,H16,H17',
scmask2=':2.C15,C16,C17,C18,C19,N4,H14,H15,H16,F1',
clambda=0.43738,
&end
/
*Also, I affixed the tleap input file for creating dual-topology structures
for complex and ligand.*
*leap.in <http://leap.in/>*
source leaprc.protein.ff14SB
source leaprc.gaff
source leaprc.water.tip3p
loadamberprep L4O.prepi
loadamberprep L4J.prepi
loadamberparams L4O.frcmod
loadamberparams L4J.frcmod
l1=loadpdb L4J.pdb
l2=loadpdb L4O.pdb
p=loadpdb Bace_protein_h.pdb
com=combine {l1 l2 p}
lig=combine {l1 l2}
set default nocenter on
solvateoct lig TIP3PBOX 15.0
addions lig Cl- 1
savepdb lig ligands_vdw_bonded.pdb
saveamberparm lig ligands_vdw_bonded.prmtop ligands_vdw_bonded.inpcrd
solvateoct com TIP3PBOX 15.0
addions com Cl- 0
addions com Na+ 10
charge com
savepdb com complex_vdw_bonded.pdb
saveamberparm com complex_vdw_bonded.prmtop complex_vdw_bonded.inpcrd
quit
Thank you so much for reading this!
Best,
Fanyu Zhao
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
