Date: Fri, 23 Sep 2022 01:33:18 +0300
Hi Robin,
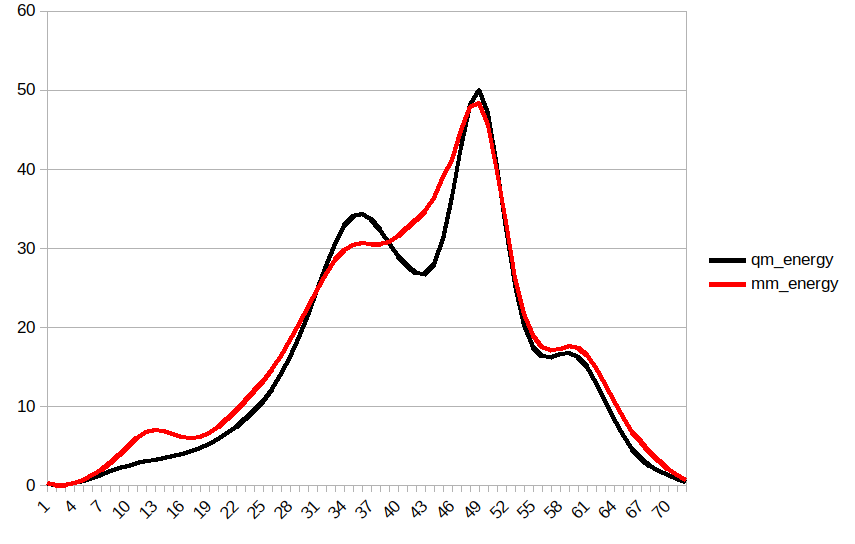
Thank you for responding. Here is the optimal MM-PES curve I got based on
my trials on many sander jobs.
[image: image.png]
Now, it is about time to fit the MM-PES curve (red in the figure) to QM-PES
curve (black in the figure)
I know that possibly I would not be able to get a similar MM-PES curve
using paramfit as I think paramfit uses some default parameters to run
minimization jobs.
Therefore, I would like to re-iterate my previous question .
Is it possible to use the MM-PES and QM-PES energies (shown in the figure
above) as inputs (BOTH OF THEM !!) in paramfit and ask paramfit to fit the
MM-PES energies to the QM-PES energies??
Cheers,
Cenk
On Thu, Sep 22, 2022 at 10:31 PM Robin Betz <robin.robinbetz.com> wrote:
> Hi Cenk,
>
> Your observation that paramfit returns better results for input structures
> that are minimized agrees with my experiences.
>
> Forcefields are a newtonian approximation of reality (which is quantum),
> and when generating terms with paramfit or similar, you want to give the
> most realistic or representative input structures in order to get the best
> possible approximation. That way, the output simulations are in the same
> broad region of conformational space that the parameters came from. To give
> a concrete example, if I fit a dihedral to only minimized structures with a
> dihedral angle of 0-5, I wouldn't expect the resulting parameters to be
> accurate for dihedral angles 90-120.
>
> Similarly, the "unphysical" structures like those generated by PES without
> minimization have really high potential energies due to steric clashing,
> electrostatics, etc. I'd guess the really high barriers you see in a PES
> calculation with sander are an example of being out of the realm of more
> realistic structures the forcefield was derived from / intended to
> describe. Fitting these structures with paramfit can also result in odd
> dihedral terms, as the program is only given a few variables it can alter
> to get the energies to match, and often will end up with bizarre values for
> dihedral potentials as a result. Under the hood it's a simple minimization
> algorithm- "change these dihedral terms such that the energies of these
> structures match the QM numbers" is the only direction, with some minor
> bounds checking but no physics knowledge.
>
> 1) Are there any parameter settings in paramfit for a short restrained (to
>> a dihedral angle of interest) minimization before fitting? For instance;
>> it would be really helpful if paramfit could restrain a dihedral of
>> interest, then run a short minimization (like 10 cycles or user-defined
>> cycles) for snapshots of the structure in a trajectory file, then
>> determine potential energies and finally fit the MM-generated PES curve to
>> QM-generated PES curve.
>
>
> At some point I had some scripting to do a restrained minimization with
> sander when generating the input structures. Conformer generation is
> definitely the most user-unfriendly part of the fitting process, and I
> agree adding that functionality to paramfit would be an improvement for the
> user experience. Unfortunately I'm pretty busy with my day job, but if
> someone wanted to contribute a script (could easily be an add-on to
> gen_conformers.sh in the paramfit tutorial) I could incorporate it into the
> docs.
>
> 2) Is there any option in paramfit to skip the step to generate a PES
>> curve by paramfit, and rather take sander (or any MM program) generated
>> PES
>> energies as well QM generated PES energies as inputs and fit them.
>
>
> You can pass any energy values to paramfit for use as the "true values" to
> be fit to. If you wanted to fit to MM energies, you could, but I'm not sure
> what the use case would be. Paramfit uses the same potential energy
> calculation as sander (the code is basically duplicated). If you're seeing
> differences between paramfit's internal energy values and sander, please do
> send me the input files as that's a bug, unless you're specifying implicit
> solvent or other additional arguments to sander.
>
> Hope this helps,
> Robin
>
>
> On Thu, Sep 22, 2022 at 10:55 AM Cenk Andac via AMBER <amber.ambermd.org>
> wrote:
>
>> Dear Amber users,
>> I have been using paramfit to fit PES curves generated by MM and QM.
>> I had posted my results in the following thread on AMBER Archive.
>> http://archive.ambermd.org/202209/0065.html
>>
>> When I implement a single-point PES run by sander, I get energy barriers
>> much higher than the corresponding energy barriers generated by QM
>> computations, which seem very similar to the energy barriers in a PES
>> curve
>> generated by paramfit as seen in the first figure in the thread above.
>> On the other hand, when I carry out a short restrained minimization
>> (restrained to a dihedral of interest) for snapshot coordinates of a
>> trajectory generated by 360 degree rotations (at 5 degree increments)
>> about
>> a dihedral angle, then I am able to get an MM-generated PES curve that
>> looks similar to a QM-generated PES curve.
>>
>> Thus, I have two questions here (or the paramfit developers could take my
>> questions as a request)
>> 1) Are there any parameter settings in paramfit for a short restrained
>> (to
>> a dihedral angle of interest) minimization before fitting? For instance;
>> it would be really helpful if paramfit could restrain a dihedral of
>> interest, then run a short minimization (like 10 cycles or user-defined
>> cycles) for snapshots of the structure in a trajectory file, then
>> determine potential energies and finally fit the MM-generated PES curve to
>> QM-generated PES curve.
>>
>> 2) Is there any option in paramfit to skip the step to generate a PES
>> curve by paramfit, and rather take sander (or any MM program) generated
>> PES
>> energies as well QM generated PES energies as inputs and fit them.
>>
>> best regards,
>>
>> Cenk Andac,
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
