Date: Thu, 15 Sep 2022 23:41:11 +0300
Dear Amber users,
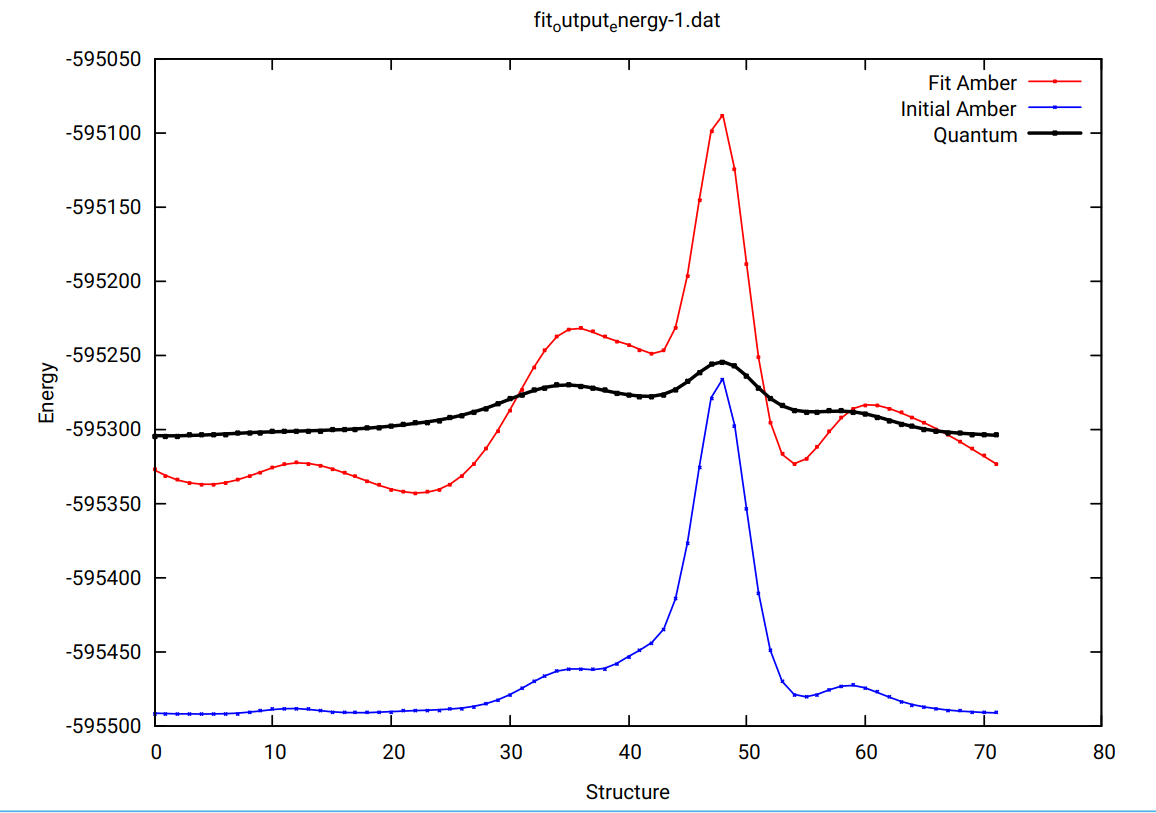
I applied paramfit to fit single-point rotational energies about a dihedral
angle (of an nt derivative) determined by MM (blue plot in the figure),in
which Vn values for the dihedral were zeroed in the force field, to single
point QM energies (at the MP2 level) for the rotations about the same
dihedral angle (black in the figure). The red curve in the figure
represents the fit energies.
I used Shaw's RNA force field for parameterization.
I see that the energies obtained by MM is about 4 times as much as energies
obtained by QM.
[image: image.png]
My question here is WHY?
Shouldn't they be about the same?
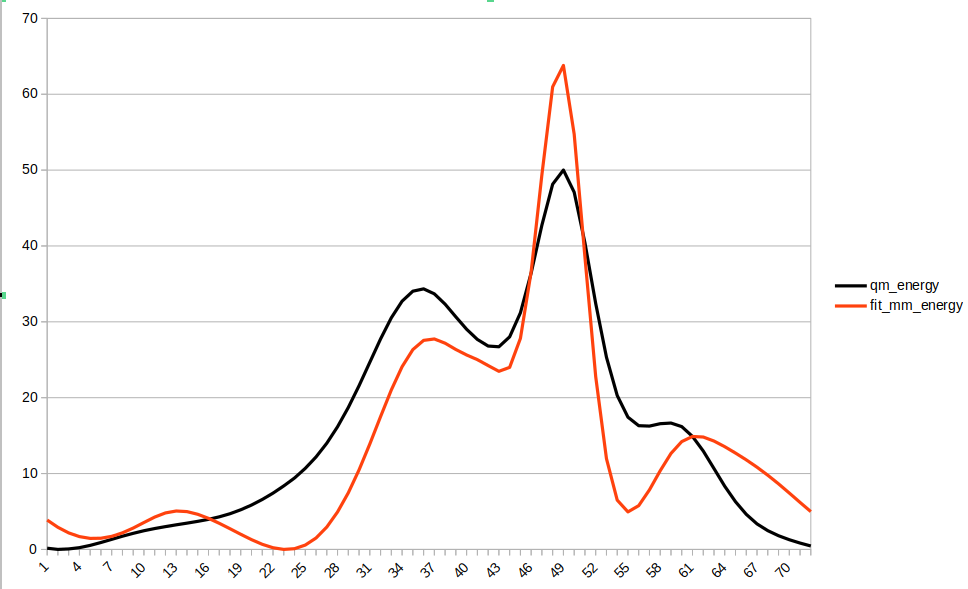
Please see the plot below after dividing the MM energies by 4. Any ideas?
[image: image.png]
best
Cenk
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
