Date: Sat, 4 Dec 2021 17:56:43 +0300
Hello,
I am trying to parameterize a metal dihedral. I tried to parameterize this
dihedral by using mdgx and/or paramfit but I couldn't achieve it. Then I
wrote a script for this but also this script failed (even if it worked
perfectly for other dihedrals), I changed the molecule design (used similar
molecules) and tried again and again but I got nothing. Here is the
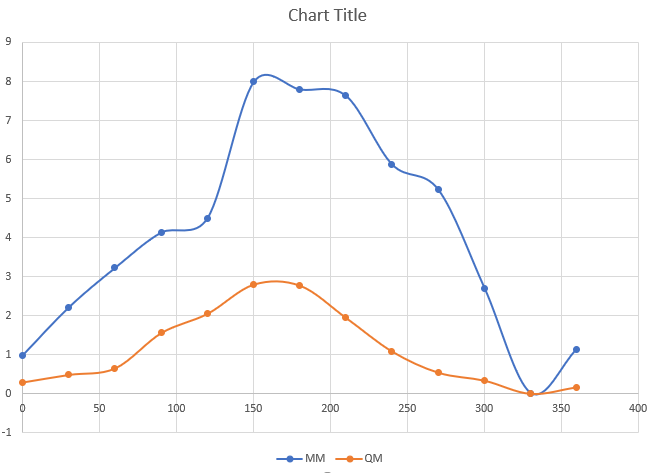
problem: Me is a metal atom and for "os-Me-ca-ca" dihedral MM relative
energy is greater than QM energy even if I put in frcmod file this:
os-Me-ca-ca 1 0.000 0.000 0.000
NOTE: bond and angle parameters are almost OK. There was no imaginer peak
in FTIR but The metal is transition metal and VFFDT calculate some
Me-os bonds as 0.000kcal/mol.
Here is the chart when I use Vn=0:
How can I solve this problem?
Thank you..
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
