Date: Thu, 11 Nov 2021 13:51:34 +0000
Dear Amber Users,
My simulation system contains isopeptide bond between
lysine and terminal glycine. I want to generate ff parameters for
isopeptide bond using PyRED. I am following tutorials at
https://upjv.q4md-forcefieldtools.org/Tutorial/Tutorial-4.php#9.
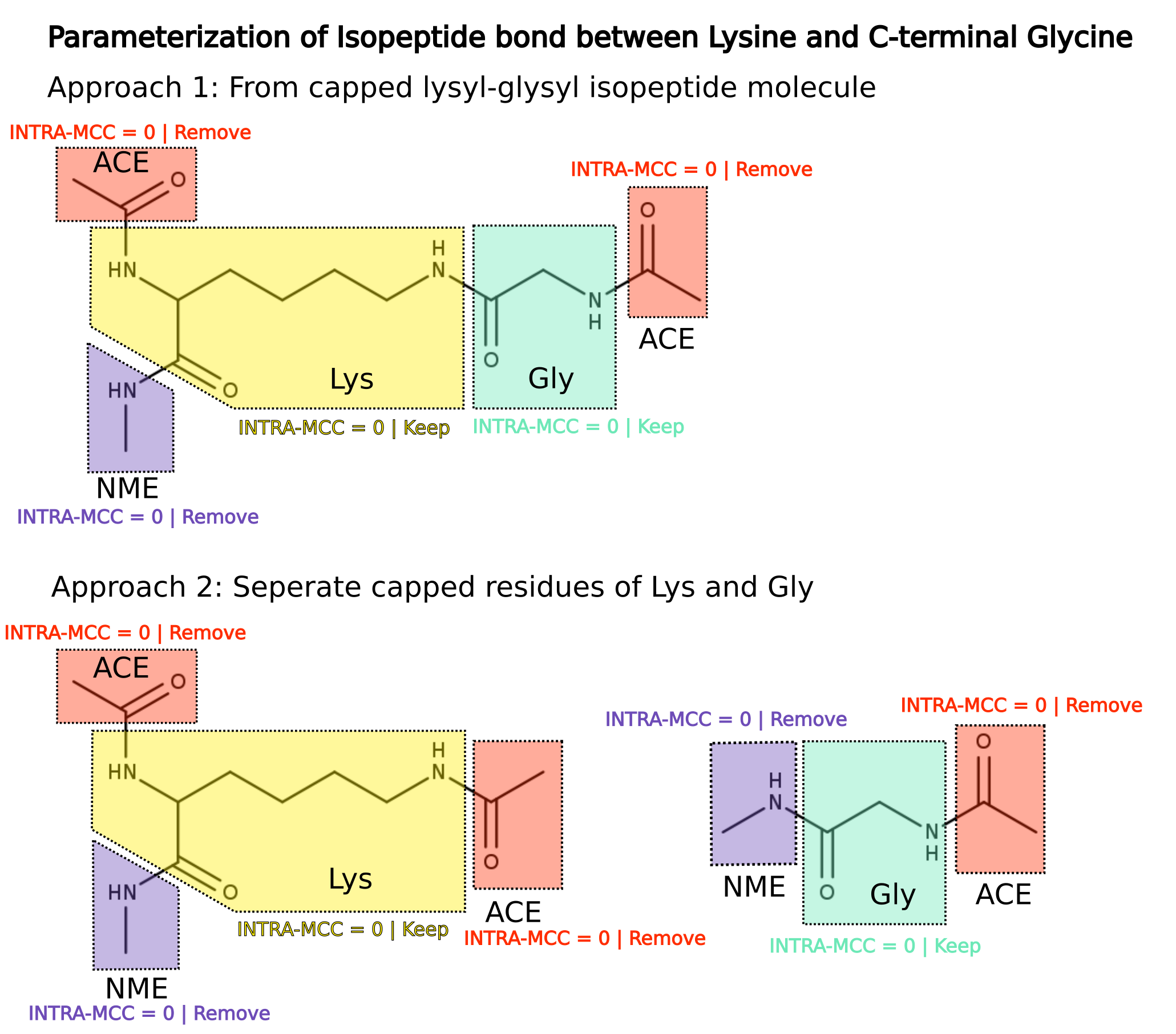
I am following two approaches (please find the schematic attached).
Approach 1: Using lysine-glycine isopeptide bonded molecule
with caps (ACE and NME). During charge fitting, charge constraints
are applied (MOLECULE1-INTRA-MCC = 0) for ACE, NME, LYS part
and GLY part and ACE and NME caps are discarded. Library for LYS and GLY parts are
saved separately (say LYX.off and GLX.off) and used during system build
with tleap.
Approach 2: ff parameters are generated from separate LYS and GLY residues
with ACE and NME caps and charge constraints are applied as in 'Approach 1'.
Please help me with the following questions:
1. What is the best between the two approaches. If both are wrong, please
2. suggest me the right approach.
3. How to choose the conformations of an input molecule (.pdb) for PyRED input.
4. I have chosen the one that is found in my pdb file.
5. What is the best charge model for charge derivation.
6. I am using RESP-A1 (HF/6-31G(d)//HF/6-31G(d) ), the default set.
7. If anyone has ff parameters available for isopeptide bond,
8. I request the same.
Thank you,
Venkat
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ISP_scheme.png)
