Date: Sat, 30 Oct 2021 07:10:22 +0500
Dear Amber users
I am working on a protein-ligand system for which I run 100 ns of
simulation. On visual inspection I have observed that ligand adopts
unexpected conformations and as shown in pdb structures. I have used RESP
charges and antechamber for calculation of parameters.
Could you please help me to figure out the source of the problem.
Thanks in advance.
Regards
Sadaf
<https://www.avast.com/sig-email?utm_medium=email&utm_source=link&utm_campaign=sig-email&utm_content=webmail>
Virus-free.
www.avast.com
<https://www.avast.com/sig-email?utm_medium=email&utm_source=link&utm_campaign=sig-email&utm_content=webmail>
<#DAB4FAD8-2DD7-40BB-A1B8-4E2AA1F9FDF2>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
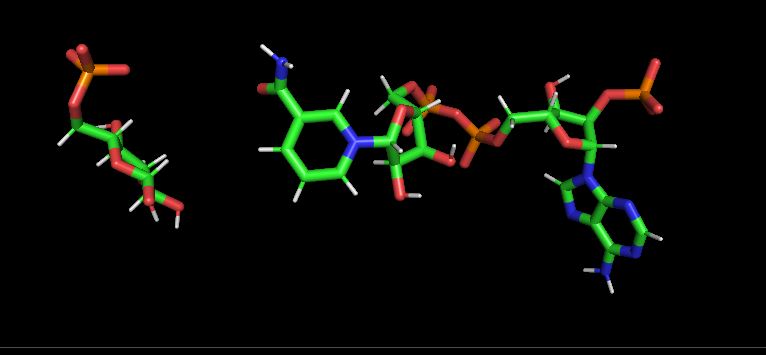
(image/jpeg attachment: ligand_conformation_at_frame_1.JPG)
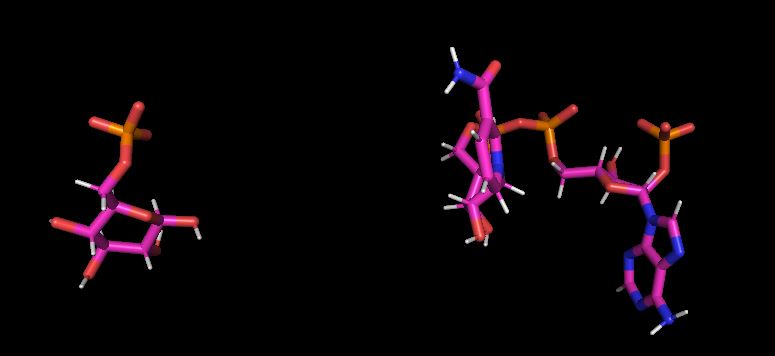
(image/jpeg attachment: ligand_conformation_at_frame_100.JPG)
