Date: Mon, 22 Feb 2021 19:27:27 +0530
Dear Amber experts,
I am using antechamber to generate GAFF parameters for a ligand
molecule. I have used the following command to do that.
antechamber -fi mol2 -fo mol2 -i ligand.mol2 -o ligand_clean_h.mol2 -rn LIG
-c bcc -pf y -at gaff2 -ek "maxcyc=0, ndiis_attempts=700"
output:
Welcome to antechamber 19.0: molecular input file processor.
acdoctor mode is on: check and diagnose problems in the input file.
-- Check Format for mol2 File --
Status: pass
-- Check Unusual Elements --
Status: pass
-- Check Open Valences --
Warning: The number of bonds (3) for atom (ID: 31, Name: N) does not match
the connectivity (2) for atom type (N.2) defined in
CORR_NAME_TYPE.DAT.
But, you may safely ignore the warnings if your molecule
uses atom names or element names as atom types.
-- Check Geometry --
for those bonded
for those not bonded
Status: pass
-- Check Weird Bonds --
/home/divya/softwares/anaconda3/bin/to_be_dispatched/antechamber: Fatal
Error!
Weird atomic valence (5) for atom (ID: 31, Name: N).
Please check atomic connectivity.
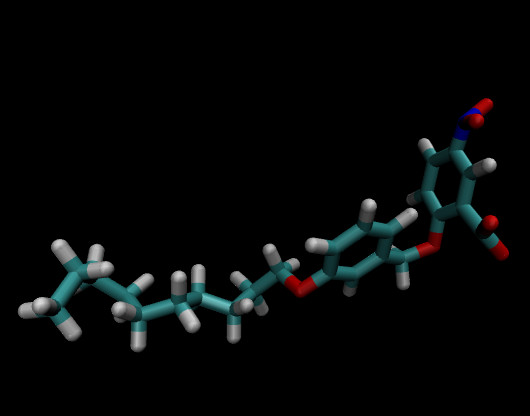
I opened the pdb file in vmd, the structure seems good, the problem I am
facing here is due to automatic bond order, and the image of the molecule
is attached here.
Please help me to figure out the problem.
Thanks,
Divya.
Dr. Divya Bharathi,
Post Doctoral Fellow,
IIIT
Hyderabad-500032.
INDIA.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: 911_5hn9.jpg)
