Date: Mon, 14 Dec 2020 19:01:40 +0530
Hi Vaibhav
I guess the error was because of the commenting the "else" line.
I have edited mdread2.F90 and attached here.
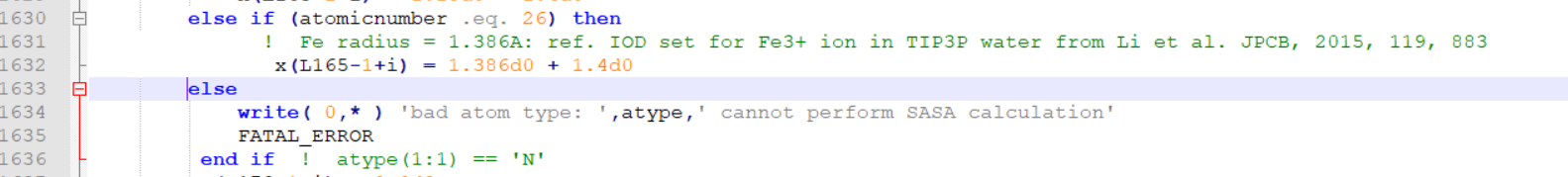
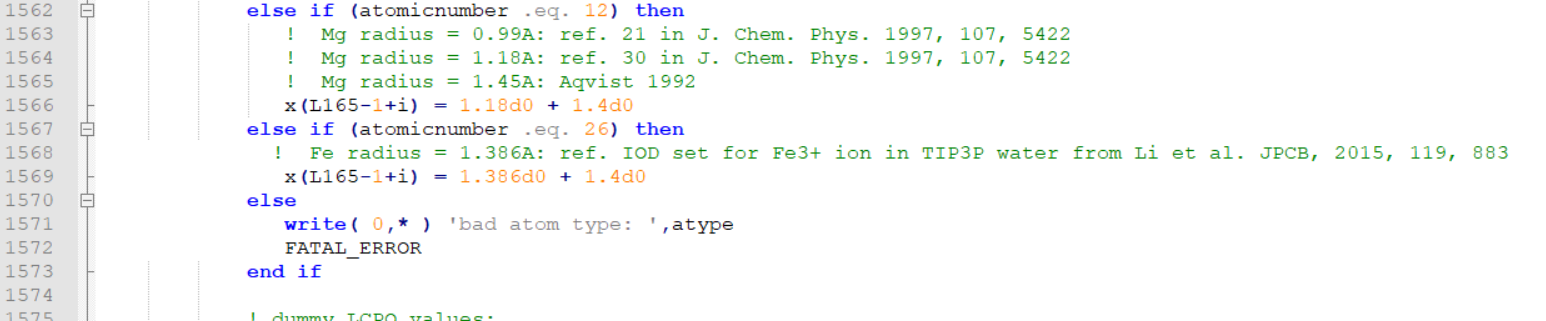
I am pasting the snapshots of the changes I did at two places.
[image: image.png]
[image: image.png]
You can directly paste this mread2.F90 in the src/sander and recompile.
Best Regards
Elvis
On Mon, 14 Dec 2020 at 18:32, Vaibhav Dixit <vaibhavadixit.gmail.com> wrote:
> Dear Elvis,
> I searched for the igb == 2 and then "bad atom type".
> I commented out the following lines (as shown).
> these are line number 1567, 1568 and 1569 in the attached file.
>
> ! else
> ! write( 0,* ) 'bad atom type: ',atype
> ! FATAL_ERROR
>
> Then added the following lines
> else if (atomicnumber .eq. 23) then
> ! Fe radius = 1.386A: ref. IOD set for Fe3+ ion in TIP3P
> water from Li et al. JPCB, 2015, 119, 883
> x(L165-1+i) = 1.386d0 + 1.4d0
>
> These line numbers are 1570, 1571 and 1572 in the attached file.
>
> Please let me know if I need to make additional changes in that file.
> Thank you and best regards.
>
>
> On Mon, Dec 14, 2020 at 9:53 PM Elvis Martis <elvis_bcp.elvismartis.in>
> wrote:
>
> > Can you point out what changes you did exactly?
> >
> >
> > Best Regards
> > Elvis
> >
> >
> >
> > On Mon, 14 Dec 2020 at 18:14, Vaibhav Dixit <vaibhavadixit.gmail.com>
> > wrote:
> >
> > > Dear All and Elivs,
> > > OK, now I got the file in amber20_src/AmberTools/src/sander.
> > > I have made the following changes (in bold), and then set DMPI and
> DCUDA
> > > both =FALSE and then ran run_make script.
> > > But I get the error shown at the end.
> > > Can you please check and help me understand what corrections to me to
> > these
> > > lines?
> > >
> > > Thanks a lot and best regards.
> > >
> > >
> > > x(L165-1+i) = 1.10d0 + 1.4d0
> > > else if (atomicnumber .eq. 12) then
> > > ! Mg radius = 0.99A: ref. 21 in J. Chem. Phys. 1997,
> 107,
> > > 5422
> > > ! Mg radius = 1.18A: ref. 30 in J. Chem. Phys. 1997,
> 107,
> > > 5422
> > > ! Mg radius = 1.45A: Aqvist 1992
> > > x(L165-1+i) = 1.18d0 + 1.4d0
> > >
> > >
> > > *# else# write( 0,* ) 'bad atom type: ',atype#
> > > FATAL_ERROR*
> > >
> > >
> > > * else if (atomicnumber .eq. 23) then ! Fe
> > radius
> > > = 1.386A: ref. IOD set for Fe3+ ion in TIP3P water from Li et al. JPCB,
> > > 2015, 119, 883 x(L165-1+i) = 1.386d0 + 1.4d0*
> > > end if
> > >
> > >
> > > *Error with run_cmake script*
> > > (base) [exx.c107739 build]$ gedit run_cmake &
> > > [2] 16544
> > > (base) [exx.c107739 build]$ make install
> > > [ 0%] Built target ucpp
> > > [ 0%] Built target fftw3_threads_obj
> > > [ 1%] Built target fftw_api
> > > [ 1%] Built target dft
> > > [ 1%] Built target dft_scalar
> > > [ 2%] Built target dft_scalar_codelets
> > > [ 5%] Built target dft_sse2_codelets
> > > [ 5%] Built target rdft
> > > [ 5%] Built target rdft_scalar
> > > [ 7%] Built target rdft_scalar_r2cb
> > > [ 8%] Built target rdft_scalar_r2cf
> > > [ 8%] Built target rdft_scalar_r2r
> > > [ 8%] Built target rdft_sse2_codelets
> > > [ 9%] Built target fftw_kernel
> > > [ 9%] Built target reodft
> > > [ 9%] Built target simd_support
> > > [ 9%] Built target fftw
> > > [ 9%] Built target libfftw_bench
> > > [ 9%] Built target fftw_benchmark_common_obj
> > > [ 9%] Built target fftw_benchmark
> > > [ 9%] Built target fftw_wisdom
> > > [ 9%] Built target fftw_mpi
> > > [ 9%] Built target fftw_mpi_benchmark
> > > [ 9%] Built target dispatch
> > > [ 9%] Built target netcdf3
> > > [ 9%] Built target netcdf
> > > [ 10%] Built target ncgen
> > > [ 10%] Built target ncgen3
> > > [ 10%] Built target nccopy
> > > [ 10%] Built target ncdump
> > > [ 11%] Built target netcdff
> > > [ 11%] Built target xblas_build
> > > [ 12%] Built target blas
> > > [ 15%] Built target lapack
> > > [ 16%] Built target arpack
> > > [ 19%] Built target pnetcdf_fortran_obj
> > > [ 19%] Built target pnetcdf_c_obj
> > > [ 19%] Built target pnetcdf
> > > [ 19%] Built target ncvalid
> > > [ 19%] Built target boost_build
> > > [ 19%] Built target amber_common
> > > [ 20%] Built target rism
> > > [ 20%] Built target libpbsa
> > > [ 20%] Built target sff_rism_interface
> > > [ 20%] Built target sff_lex
> > > [ 20%] Built target dsarpack_obj
> > > [ 20%] Built target sff
> > > [ 21%] Built target gbnsr6
> > > [ 21%] Built target cifparse
> > > [ 22%] Built target addles
> > > [ 23%] Built target rism_mpi
> > > [ 23%] Built target sander_rism_interface
> > > [ 25%] Built target sqm_common
> > > [ 25%] Built target libsqm
> > > [ 26%] Built target sff_fortran
> > > [ 26%] Building Fortran object
> > > AmberTools/src/sander/CMakeFiles/sander_base_obj.dir/mdread.F90.o
> > >
> /home/exx/Downloads/amber20_src/AmberTools/src/sander/mdread2.F90:1571.1:
> > > Included at
> > > /home/exx/Downloads/amber20_src/AmberTools/src/sander/mdread.F90:24:
> > >
> > > \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0!
> > \xC2\xA0Fe
> > > rad
> > > 1
> > > Error: Invalid character in name at (1)
> > >
> /home/exx/Downloads/amber20_src/AmberTools/src/sander/mdread2.F90:1572.1:
> > > Included at
> > > /home/exx/Downloads/amber20_src/AmberTools/src/sander/mdread.F90:24:
> > >
> > > \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0 \xC2\xA0
> > x(L165-1+i)
> > > = 1
> > > 1
> > > Error: Invalid character in name at (1)
> > > make[2]: ***
> > > [AmberTools/src/sander/CMakeFiles/sander_base_obj.dir/mdread.F90.o]
> > Error 1
> > > make[1]: *** [AmberTools/src/sander/CMakeFiles/sander_base_obj.dir/all]
> > > Error 2
> > > make: *** [all] Error 2
> > > [2]+ Done gedit run_cmake
> > >
> > > On Mon, Dec 14, 2020 at 8:36 PM Elvis Martis <elvis_bcp.elvismartis.in
> >
> > > wrote:
> > >
> > > > One more thing I missed,
> > > > After you have successfully located the file and edited it
> accordingly,
> > > > please don't forget to recompile AMBER.
> > > > Best Regards
> > > > Elvis
> > > >
> > > >
> > > >
> > > > On Mon, 14 Dec 2020 at 17:05, Elvis Martis <elvis_bcp.elvismartis.in
> >
> > > > wrote:
> > > >
> > > > > OK.
> > > > > Have you compiled AMBER20 using the legacy configure method or the
> > new
> > > > > cmake method?
> > > > > If you have using the new cmake method of compilation, then I guess
> > you
> > > > > should find that file in
> > amber20_src/AmberTools/src/sander/mdread2.F90.
> > > > > if you have compiled using the legacy method, then you files should
> > in
> > > > the
> > > > > path where I sent previously.
> > > > > Best Regards
> > > > > Elvis
> > > > >
> > > > >
> > > > >
> > > > > On Mon, 14 Dec 2020 at 16:55, Vaibhav Dixit <
> vaibhavadixit.gmail.com
> > >
> > > > > wrote:
> > > > >
> > > > >> Dear All and Elivs,
> > > > >> I'm afraid, I still can't find this file inside the AmberTools
> > folder,
> > > > >> there is no folder by the same sander.
> > > > >> All tests passed while installing Amber20, thus I think there is
> > some
> > > > >> confusion regarding the directory structure and file locations in
> > > > Amber18
> > > > >> and Amber20 as well.
> > > > >> Please let me know if you know the fix to this as well.
> > > > >> thank you and best regards.
> > > > >>
> > > > >> (base) [exx.c107739 amber20]$ ls
> > > > >> $AMBERHOME/AmberTools/src/sander/mdread2.F90
> > > > >> ls: cannot access
> > > > >> /home/exx/Downloads/amber20/AmberTools/src/sander/mdread2.F90: No
> > such
> > > > >> file
> > > > >> or directory
> > > > >> (base) [exx.c107739 amber20]$ cd AmberTools/src/
> > > > >> (base) [exx.c107739 src]$ ls -arlth
> > > > >> total 28K
> > > > >> drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:17 cpptraj
> > > > >> drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:17 mm_pbsa
> > > > >> drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:17 FEW
> > > > >> drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:17 pytraj
> > > > >> drwxrwxr-x. 7 exx exx 4.0K Dec 4 15:25 ..
> > > > >> -rw-r--r--. 1 exx exx 1.9K Dec 4 17:26 config.h
> > > > >> drwxrwxr-x. 6 exx exx 4.0K Dec 4 17:45 .
> > > > >>
> > > > >> On Mon, Dec 14, 2020 at 7:46 PM Elvis Martis <
> > > elvis_bcp.elvismartis.in>
> > > > >> wrote:
> > > > >>
> > > > >> > Hi
> > > > >> > Sorry my bad. For AMBER20
> > > > >> > you can look in
> > > > >> > $AMBERHOME/AmberTools/src/sander/mdread2.F90
> > > > >> > Start looking from line number 1508 (this should be same unless
> > > there
> > > > >> were
> > > > >> > changes made earlier)
> > > > >> > this line starts with " else if ( gbsa == 2 ) then"
> > > > >> >
> > > > >> > Best Regards
> > > > >> > Elvis
> > > > >> >
> > > > >> >
> > > > >> >
> > > > >> > On Mon, 14 Dec 2020 at 15:57, Vaibhav Dixit <
> > > vaibhavadixit.gmail.com>
> > > > >> > wrote:
> > > > >> >
> > > > >> > > Dear All,
> > > > >> > > I got this suggestion earlier last month also.
> > > > >> > > But to my surprise, I can't find these specified folders and
> > files
> > > > in
> > > > >> the
> > > > >> > > expected locations.
> > > > >> > > I ran a find command to search for the mdread.f file, but it
> is
> > > not
> > > > >> found
> > > > >> > > anywhere inside amber20 folder and subfolders.
> > > > >> > > This command works and finds other files as shown below.
> > > > >> > >
> > > > >> > > Please do let me know what I'm missing to understand here.
> > > > >> > > thanks
> > > > >> > >
> > > > >> > > (base) [exx.c107739 amber20]$ ls -arlth
> > > > >> > > total 128K
> > > > >> > > -rw-r--r--. 1 exx exx 7.5K Dec 6 2018 GNU_LGPL_v3
> > > > >> > > -rwxr-xr-x. 1 exx exx 116 Dec 6 2018 amber-interactive.sh
> > > > >> > > -rw-r--r--. 1 exx exx 1.1K Apr 28 2020 Makefile
> > > > >> > > -rw-r--r--. 1 exx exx 37K Apr 28 2020 LICENSE
> > > > >> > > drwxrwxr-x. 8 exx exx 4.0K Dec 4 15:17 licenses
> > > > >> > > drwxrwxr-x. 2 exx exx 4.0K Dec 4 15:17 doc
> > > > >> > > drwxrwxr-x. 19 exx exx 4.0K Dec 4 15:17 miniconda
> > > > >> > > drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:17 share
> > > > >> > > drwxrwxr-x. 9 exx exx 4.0K Dec 4 15:17 benchmarks
> > > > >> > > drwxrwxr-x. 16 exx exx 4.0K Dec 4 15:17 dat
> > > > >> > > drwxrwxr-x. 7 exx exx 4.0K Dec 4 15:25 AmberTools
> > > > >> > > -rw-r--r--. 1 exx exx 1.9K Dec 4 17:26 config.h
> > > > >> > > drwxrwxr-x. 8 exx exx 4.0K Dec 10 15:54 logs
> > > > >> > > drwxrwxr-x. 157 exx exx 4.0K Dec 10 15:58 test
> > > > >> > > -rwxr-xr-x. 1 exx exx 1.8K Dec 10 20:38 amber.sh
> > > > >> > > -rwxr-xr-x. 1 exx exx 1.9K Dec 10 20:38 amber.csh
> > > > >> > > drwxrwxr-x. 15 exx exx 4.0K Dec 10 21:50 .
> > > > >> > > drwxr-xr-x. 2 exx exx 4.0K Dec 10 21:50 include
> > > > >> > > drwxrwxr-x. 4 exx exx 4.0K Dec 10 21:50 lib64
> > > > >> > > drwxr-xr-x. 4 exx exx 4.0K Dec 10 21:50 lib
> > > > >> > > drwxrwxr-x. 3 exx exx 4.0K Dec 10 21:50 bin
> > > > >> > > drwxr-xr-x. 24 exx exx 4.0K Dec 12 12:13 ..
> > > > >> > > (base) [exx.c107739 amber20]$ cd AmberTools/
> > > > >> > > (base) [exx.c107739 AmberTools]$ ls -arlth
> > > > >> > > total 28K
> > > > >> > > drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:17 benchmarks
> > > > >> > > drwxrwxr-x. 6 exx exx 4.0K Dec 4 15:17 examples
> > > > >> > > drwxrwxr-x. 3 exx exx 4.0K Dec 4 15:25 .pytest_cache
> > > > >> > > drwxrwxr-x. 7 exx exx 4.0K Dec 4 15:25 .
> > > > >> > > drwxrwxr-x. 6 exx exx 4.0K Dec 4 17:45 src
> > > > >> > > drwxrwxr-x. 15 exx exx 4.0K Dec 10 21:50 ..
> > > > >> > > drwxrwxr-x. 65 exx exx 4.0K Dec 10 21:59 test
> > > > >> > > (base) [exx.c107739 AmberTools]$ cd ..
> > > > >> > > (base) [exx.c107739 amber20]$ find . -name mdread.f
> > > > >> > > (base) [exx.c107739 amber20]$ find . -name amber.sh
> > > > >> > > ./amber.sh
> > > > >> > > (base) [exx.c107739 amber20]$ find . -name am1bcc
> > > > >> > > ./bin/am1bcc
> > > > >> > > ./bin/wrapped_progs/am1bcc
> > > > >> > >
> > > > >> > > On Mon, Dec 14, 2020 at 7:14 PM Elvis Martis <
> > > > >> elvis_bcp.elvismartis.in>
> > > > >> > > wrote:
> > > > >> > >
> > > > >> > > > Hi
> > > > >> > > > Here is a solution in a different thread, you will have to
> > edit
> > > > the
> > > > >> > > > mdread.f file. Please have a look at link below to know how
> to
> > > > edit
> > > > >> the
> > > > >> > > > file and where to find it.
> > > > >> > > > Re: [AMBER] bad atom type: Br from Jason Swails on
> 2011-11-11
> > > > (Amber
> > > > >> > > > Archive Nov 2011) (ambermd.org)
> > > > >> > > > <http://archive.ambermd.org/201111/0370.html>
> > > > >> > > > Best Regards
> > > > >> > > > Elvis
> > > > >> > > >
> > > > >> > > >
> > > > >> > > >
> > > > >> > > > On Mon, 14 Dec 2020 at 15:34, Vaibhav Dixit <
> > > > >> vaibhavadixit.gmail.com>
> > > > >> > > > wrote:
> > > > >> > > >
> > > > >> > > > > Dear All,
> > > > >> > > > > I'm getting the following error message while trying to
> run
> > > > >> MMPBSA.py
> > > > >> > > in
> > > > >> > > > > Amber20.
> > > > >> > > > > It says that the receptor mask overwritten with default,
> > > which I
> > > > >> > don't
> > > > >> > > > know
> > > > >> > > > > why it is doing even after I gave a specific receptor mask
> > > (1st
> > > > >> > residue
> > > > >> > > > is
> > > > >> > > > > my ligand in the trajectory). It also gives the bad atom
> > type
> > > > >> error,
> > > > >> > > > that
> > > > >> > > > > I got with Amber18 last month.
> > > > >> > > > >
> > > > >> > > > > >From the output files (attached), it is not clear what is
> > the
> > > > >> exact
> > > > >> > > > nature
> > > > >> > > > > of the error, thus I have no clue how to proceed to fix
> the
> > > > same.
> > > > >> > > > > Can you please help me understand why the dry prmtop is
> not
> > > > >> > compatible?
> > > > >> > > > > And how can I possibly attempt to get this analysis done?
> > > > >> > > > > My mmpbsa input file is given below.
> > > > >> > > > > Thanking you all in advance.
> > > > >> > > > > Best regards
> > > > >> > > > >
> > > > >> > > > > mmpbsa input file:
> > > > >> > > > > (base) [exx.c107739 mmpbsa-6701-tyr]$ more
> mmpbsa-decomp.in
> > > > >> > > > > &general
> > > > >> > > > > startframe=1, endframe=100, interval=5,
> > > > >> > > > > verbose=2, keep_files=2, netcdf=1, receptor_mask=2-386,
> > > > >> > > > > strip_mask=:387-16750,
> > > > >> > > > > /
> > > > >> > > > > &gb
> > > > >> > > > > igb=5, saltcon=0.150,
> > > > >> > > > > /
> > > > >> > > > > &pb
> > > > >> > > > > istrng=0.15, fillratio=4.0,
> > > > >> > > > > /
> > > > >> > > > > &decomp
> > > > >> > > > > idecomp=1, print_res="56-57; 74-75; 167; 222; 225-227;
> > > > 229-231;
> > > > >> > > > 272-273;
> > > > >> > > > > 275-277; 336; 368; 370; 372-373"
> > > > >> > > > > dec_verbose=1,
> > > > >> > > > > /
> > > > >> > > > >
> > > > >> > > > >
> > > > >> > > > > Error message on the terminal
> > > > >> > > > > (base) [exx.c107739 mmpbsa-6701-tyr]$ MMPBSA.py -O -i
> > > > >> > mmpbsa-decomp.in
> > > > >> > > > -o
> > > > >> > > > > mmpbs-rouf-6701-tyr.dat -do decomp-rouf-6701-tyr.dat -sp
> > > > >> > > > > ../rouf-6701-tyr-solv.prmtop -cp
> ../rouf-6701-tyr-dry.prmtop
> > > -lp
> > > > >> > > > > ../tyr-dry.prmtop -y ../rouf-6701-tyr-solv-prod300.nc -rp
> > > > >> > > > > ../rouf-6701-nolig-dry.prmtop
> > > > >> > > > > Loading and checking parameter files for compatibility...
> > > > >> > > > >
> > > > >> > > > >
> > > > >> > > >
> > > > >> > >
> > > > >> >
> > > > >>
> > > >
> > >
> >
> */home/exx/Downloads/amber20/lib/python3.8/site-packages/MMPBSA_mods/main.py:603:
> > > > >> > > > > UserWarning: receptor_mask overwritten with default*
> > > > >> > > > >
> > > > >> > > > > warnings.warn('receptor_mask overwritten with
> default\n')
> > > > >> > > > > cpptraj found! Using
> /home/exx/Downloads/amber20/bin/cpptraj
> > > > >> > > > > sander found! Using /home/exx/Downloads/amber20/bin/sander
> > > > >> > > > > Preparing trajectories for simulation...
> > > > >> > > > > 20 frames were processed by cpptraj for use in
> calculation.
> > > > >> > > > >
> > > > >> > > > > Running calculations on normal system...
> > > > >> > > > >
> > > > >> > > > > Beginning GB calculations with
> > > > >> /home/exx/Downloads/amber20/bin/sander
> > > > >> > > > > calculating complex contribution...
> > > > >> > > > > * bad atom type: M1*
> > > > >> > > > > File "/home/exx/Downloads/amber20/bin/MMPBSA.py", line
> > 100,
> > > in
> > > > >> > > <module>
> > > > >> > > > > app.run_mmpbsa()
> > > > >> > > > > File
> > > > >> > > > >
> > > > >> > > > >
> > > > >> > > >
> > > > >> > >
> > > > >> >
> > > > >>
> > > >
> > >
> >
> "/home/exx/Downloads/amber20/lib/python3.8/site-packages/MMPBSA_mods/main.py",
> > > > >> > > > > line 218, in run_mmpbsa
> > > > >> > > > > self.calc_list.run(rank, self.stdout)
> > > > >> > > > > File
> > > > >> > > > >
> > > > >> > > > >
> > > > >> > > >
> > > > >> > >
> > > > >> >
> > > > >>
> > > >
> > >
> >
> "/home/exx/Downloads/amber20/lib/python3.8/site-packages/MMPBSA_mods/calculation.py",
> > > > >> > > > > line 82, in run
> > > > >> > > > > calc.run(rank, stdout=stdout, stderr=stderr)
> > > > >> > > > > File
> > > > >> > > > >
> > > > >> > > > >
> > > > >> > > >
> > > > >> > >
> > > > >> >
> > > > >>
> > > >
> > >
> >
> "/home/exx/Downloads/amber20/lib/python3.8/site-packages/MMPBSA_mods/calculation.py",
> > > > >> > > > > line 156, in run
> > > > >> > > > > raise CalcError('%s failed with prmtop %s!' %
> > > (self.program,
> > > > >> > > > > CalcError: /home/exx/Downloads/amber20/bin/sander failed
> > with
> > > > >> prmtop
> > > > >> > > > > ../rouf-6701-tyr-dry.prmtop!
> > > > >> > > > > Exiting. All files have been retained.
> > > > >> > > > >
> > > > >> > > > > --
> > > > >> > > > >
> > > > >> > > > > Regards,
> > > > >> > > > >
> > > > >> > > > > Dr. Vaibhav A. Dixit,
> > > > >> > > > >
> > > > >> > > > > Visiting Scientist at the Manchester Institute of
> > > Biotechnology
> > > > >> > (MIB),
> > > > >> > > > The
> > > > >> > > > > University of Manchester, 131 Princess Street, Manchester
> M1
> > > > 7DN,
> > > > >> UK.
> > > > >> > > > > AND
> > > > >> > > > > Assistant Professor,
> > > > >> > > > > Department of Pharmacy,
> > > > >> > > > > ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> > > > >> > > > > Birla Institute of Technology and Sciences Pilani
> > > (BITS-Pilani),
> > > > >> > > > > VidyaVihar Campus, street number 41, Pilani, Rajasthan
> > 333031.
> > > > >> > > > > India.
> > > > >> > > > > Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> > > > >> > > > > Email: vaibhav.dixit.pilani.bits-pilani.ac.in,
> > > > >> > vaibhavadixit.gmail.com
> > > > >> > > > > http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> > > > >> > > > > https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
> > > > >> > > > >
> > > > >> > > > > ORCID ID: https://orcid.org/0000-0003-4015-2941
> > > > >> > > > >
> > > > >> > > > >
> > > > >>
> > http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
> > > > >> > > > >
> > > > >> > > > > P Please consider the environment before printing this
> > > > >> > > > > _______________________________________________
> > > > >> > > > > AMBER mailing list
> > > > >> > > > > AMBER.ambermd.org
> > > > >> > > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >> > > > >
> > > > >> > > > _______________________________________________
> > > > >> > > > AMBER mailing list
> > > > >> > > > AMBER.ambermd.org
> > > > >> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >> > > >
> > > > >> > >
> > > > >> > >
> > > > >> > > --
> > > > >> > >
> > > > >> > > Regards,
> > > > >> > >
> > > > >> > > Dr. Vaibhav A. Dixit,
> > > > >> > >
> > > > >> > > Visiting Scientist at the Manchester Institute of
> Biotechnology
> > > > (MIB),
> > > > >> > The
> > > > >> > > University of Manchester, 131 Princess Street, Manchester M1
> > 7DN,
> > > > UK.
> > > > >> > > AND
> > > > >> > > Assistant Professor,
> > > > >> > > Department of Pharmacy,
> > > > >> > > ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> > > > >> > > Birla Institute of Technology and Sciences Pilani
> (BITS-Pilani),
> > > > >> > > VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031.
> > > > >> > > India.
> > > > >> > > Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> > > > >> > > Email: vaibhav.dixit.pilani.bits-pilani.ac.in,
> > > > >> vaibhavadixit.gmail.com
> > > > >> > > http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> > > > >> > > https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
> > > > >> > >
> > > > >> > > ORCID ID: https://orcid.org/0000-0003-4015-2941
> > > > >> > >
> > > > >> > >
> > > > http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
> > > > >> > >
> > > > >> > > P Please consider the environment before printing this e-mail
> > > > >> > > _______________________________________________
> > > > >> > > AMBER mailing list
> > > > >> > > AMBER.ambermd.org
> > > > >> > > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >> > >
> > > > >> > _______________________________________________
> > > > >> > AMBER mailing list
> > > > >> > AMBER.ambermd.org
> > > > >> > http://lists.ambermd.org/mailman/listinfo/amber
> > > > >> >
> > > > >>
> > > > >>
> > > > >> --
> > > > >>
> > > > >> Regards,
> > > > >>
> > > > >> Dr. Vaibhav A. Dixit,
> > > > >>
> > > > >> Visiting Scientist at the Manchester Institute of Biotechnology
> > (MIB),
> > > > The
> > > > >> University of Manchester, 131 Princess Street, Manchester M1 7DN,
> > UK.
> > > > >> AND
> > > > >> Assistant Professor,
> > > > >> Department of Pharmacy,
> > > > >> ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> > > > >> Birla Institute of Technology and Sciences Pilani (BITS-Pilani),
> > > > >> VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031.
> > > > >> India.
> > > > >> Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> > > > >> Email: vaibhav.dixit.pilani.bits-pilani.ac.in,
> > > vaibhavadixit.gmail.com
> > > > >> http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> > > > >> https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
> > > > >>
> > > > >> ORCID ID: https://orcid.org/0000-0003-4015-2941
> > > > >>
> > > > >>
> > http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
> > > > >>
> > > > >> P Please consider the environment before printing this e-mail
> > > > >> _______________________________________________
> > > > >> AMBER mailing list
> > > > >> AMBER.ambermd.org
> > > > >> http://lists.ambermd.org/mailman/listinfo/amber
> > > > >>
> > > > >
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > >
> > >
> > > --
> > >
> > > Regards,
> > >
> > > Dr. Vaibhav A. Dixit,
> > >
> > > Visiting Scientist at the Manchester Institute of Biotechnology (MIB),
> > The
> > > University of Manchester, 131 Princess Street, Manchester M1 7DN, UK.
> > > AND
> > > Assistant Professor,
> > > Department of Pharmacy,
> > > ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> > > Birla Institute of Technology and Sciences Pilani (BITS-Pilani),
> > > VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031.
> > > India.
> > > Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> > > Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com
> > > http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> > > https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
> > >
> > > ORCID ID: https://orcid.org/0000-0003-4015-2941
> > >
> > > http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
> > >
> > > P Please consider the environment before printing this e-mail
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
>
>
> --
>
> Regards,
>
> Dr. Vaibhav A. Dixit,
>
> Visiting Scientist at the Manchester Institute of Biotechnology (MIB), The
> University of Manchester, 131 Princess Street, Manchester M1 7DN, UK.
> AND
> Assistant Professor,
> Department of Pharmacy,
> ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄
> Birla Institute of Technology and Sciences Pilani (BITS-Pilani),
> VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031.
> India.
> Phone No. +91 1596 255652, Mob. No. +91-7709129400,
> Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com
> http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile
> https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/
>
> ORCID ID: https://orcid.org/0000-0003-4015-2941
>
> http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra
>
> P Please consider the environment before printing this e-mail
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
- application/octet-stream attachment: mdread2.F90
