Date: Fri, 27 Nov 2020 17:15:49 +0000
Hello everyone,
I am running an absolute binding free energy calculation on a ABL kinase system ( ligand is isopropanol), I have applied distance based restraints to keep the ligand in the pocket:
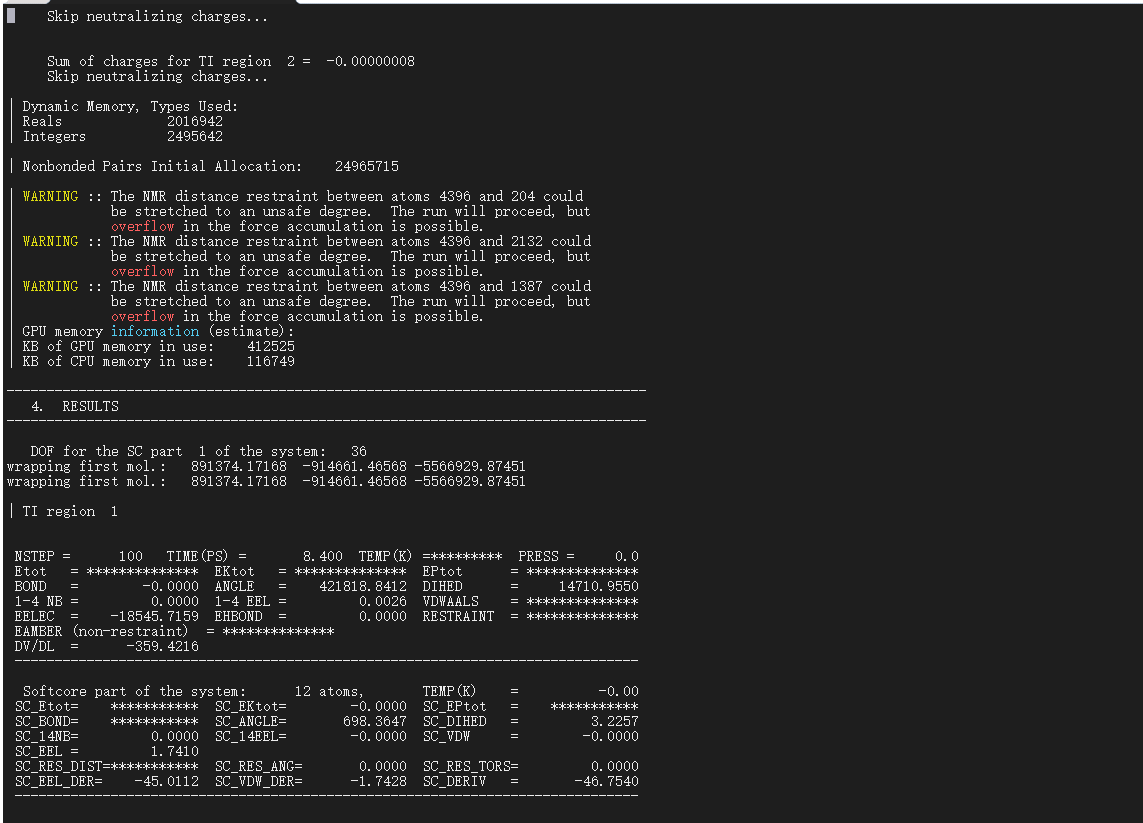
However, after minimization, the system gets unstable in the temperature ramping simulation I get this: ( see picture below). Any ideas/thoughts on how to prevent this?
I have used softcore potentials and I am doing a stepwise de-charge, then disappearing the LJ interactions.
Thanks
Debarati
[cid:image001.png.01D6C4B6.D8F0CCC0]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: C050DC057C6A44A8B852E2233E4D487A.png)
