From: 辛志宏 <xzhfood.njau.edu.cn>
Date: Fri, 13 Nov 2020 21:41:02 +0800 (GMT+08:00)
Dear all,
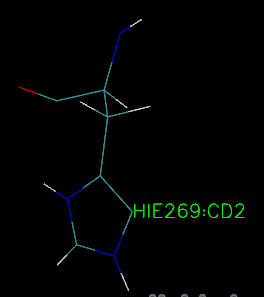
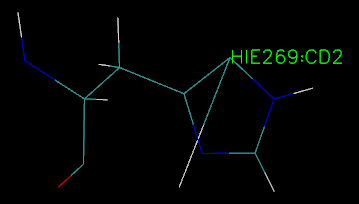
I parmetered and solvented the enzyme and saved the top and rst as well as PDB file successfully by tleap, and then open the top an rst file by VMD, it was found that
the direction of H Aatom in the reside 269 is not correct, but it is correct when I open the PDB file in the VMD, I don't know what's reason for the same enzyme in
different file style, how to correct the error from the top and rst file.
Any help for the solution will be much appreciation.
Attachment are the two pictures
Zhihong Xin,
College of Food Science and Technology
Nanjing Agricultural Univisity
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Nov 13 2020 - 06:00:04 PST