Date: Wed, 23 Sep 2020 12:19:04 +0530
Dear Amber developers/webmaster,
I observed that the main-tutorial page has been restructured since I
visited a couple of months ago. This is excellent as it looks more/better
structured now.
A great thanks to the Amber team.
Are you considering to add another section useful for self-(re)training on
the *basics of minimization, and heating steps*?
Currently, after system setup/non-standard residue, there is a section
directly on running MD.
Please redirect me to the relevant tutorial if these are already discussed
at length somewhere that I missed (I keep forgetting the imp of various
options).
I'm simulating a system where the E has not minimized even after 4-5 steps,
the E as seen with mdout_analyzer keep getting lower. Thus I think I need a
refresher in the basics, but I can't find such tutorial easily.
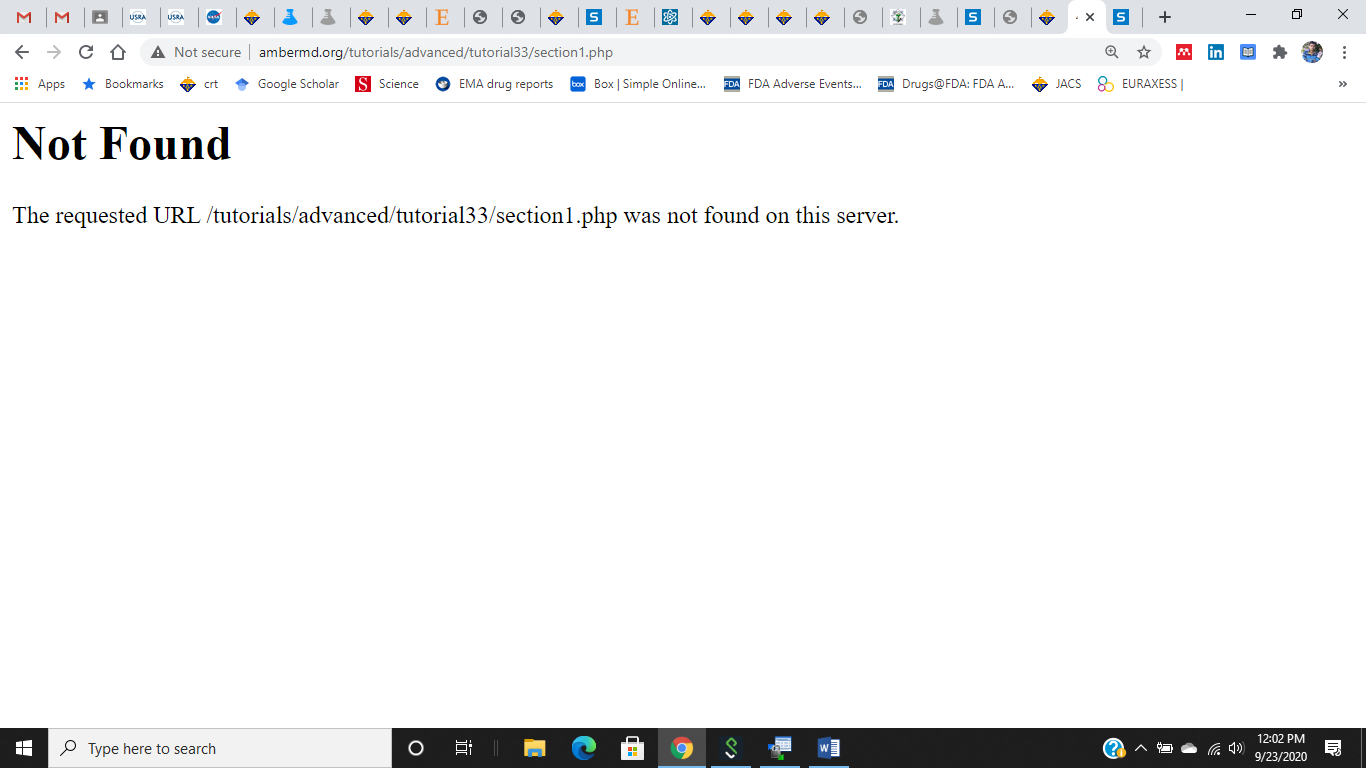
Additionally, I'm unable to access the constant pH and Redox potential
tutorial sections as shown below.
http://ambermd.org/tutorials/advanced/tutorial33/section1.php
Please redirect if there are new pages for the same.
Thank you and best regards
[image: image.png]
-- Regards, Dr. Vaibhav A. Dixit, Assistant Professor, Department of Pharmacy, ▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄▄ Birla Institute of Technology and Sciences Pilani (BITS-Pilani), VidyaVihar Campus, street number 41, Pilani, Rajasthan 333031. India. Phone No. +91 1596 255652, Mob. No. +91-7709129400, Email: vaibhav.dixit.pilani.bits-pilani.ac.in, vaibhavadixit.gmail.com http://www.bits-pilani.ac.in/pilani/vaibhavdixit/profile https://www.linkedin.com/in/vaibhav-dixit-b1a07a39/ ORCID ID: https://orcid.org/0000-0003-4015-2941 http://scholar.google.co.in/citations?user=X876BKcAAAAJ&hl=en&oi=sra P Please consider the environment before printing this e-mail
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
