Date: Tue, 8 Sep 2020 17:42:51 +0200
Dear Amber Users,
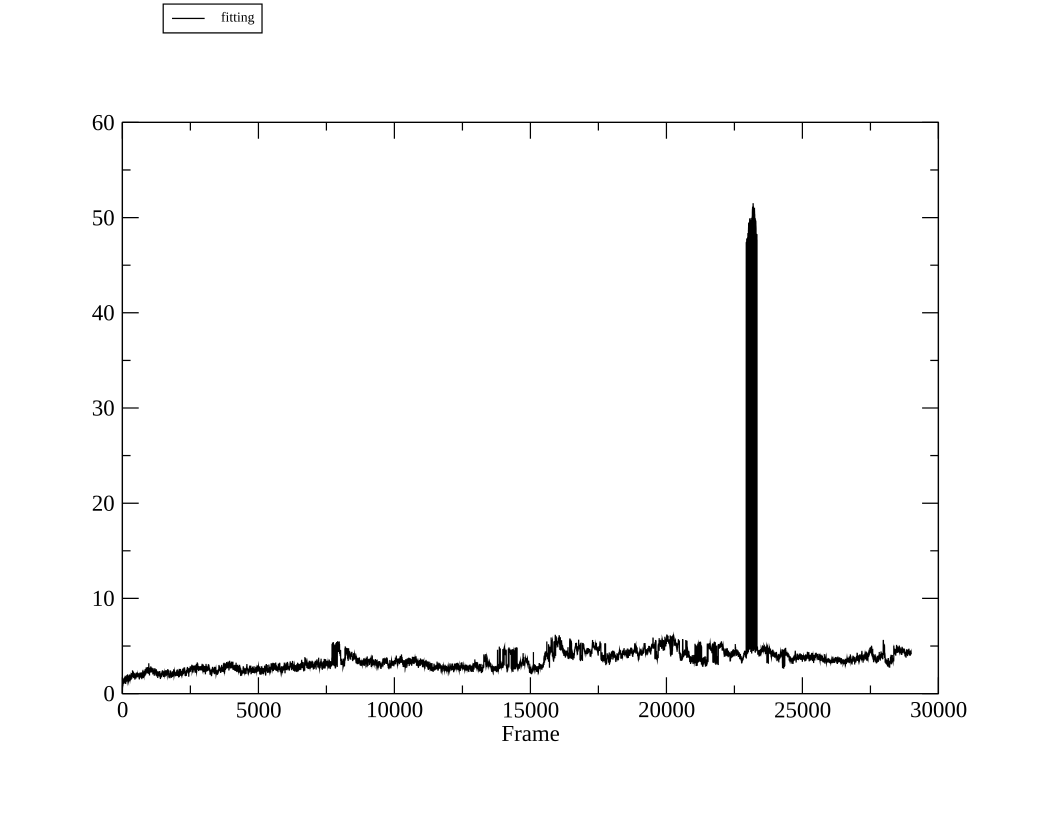
I am running a simulation of a large homodimeric protein (15800 atoms). The
simulation mostly runs well, but a large tower/spike of RMSD around 240ns.
I have tried to address the artefact by using autoimage, and anchoring to
the residues of the structure, but the issue persists.
I am using pmemd.cuda for the simulation.
Have you any advice of how to resolve this, or ideas of what could be
causing this? I will be rerunning that section of simulation in the mean
time to see if anything changes.
Warm Regards
Jorge da Rocha
-- Jorge da Rocha (MSc Med) PhD Candidate at the Sydney Brenner Institute for Molecular Bioscience Division of Human Genetics, Faculty of Health Sciences University of the Witwatersrand Cell: 082 944 1651
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: md_290ns_strange_spike.png)
