Date: Sat, 08 Aug 2020 21:58:49 +0800
Dear Amber community!
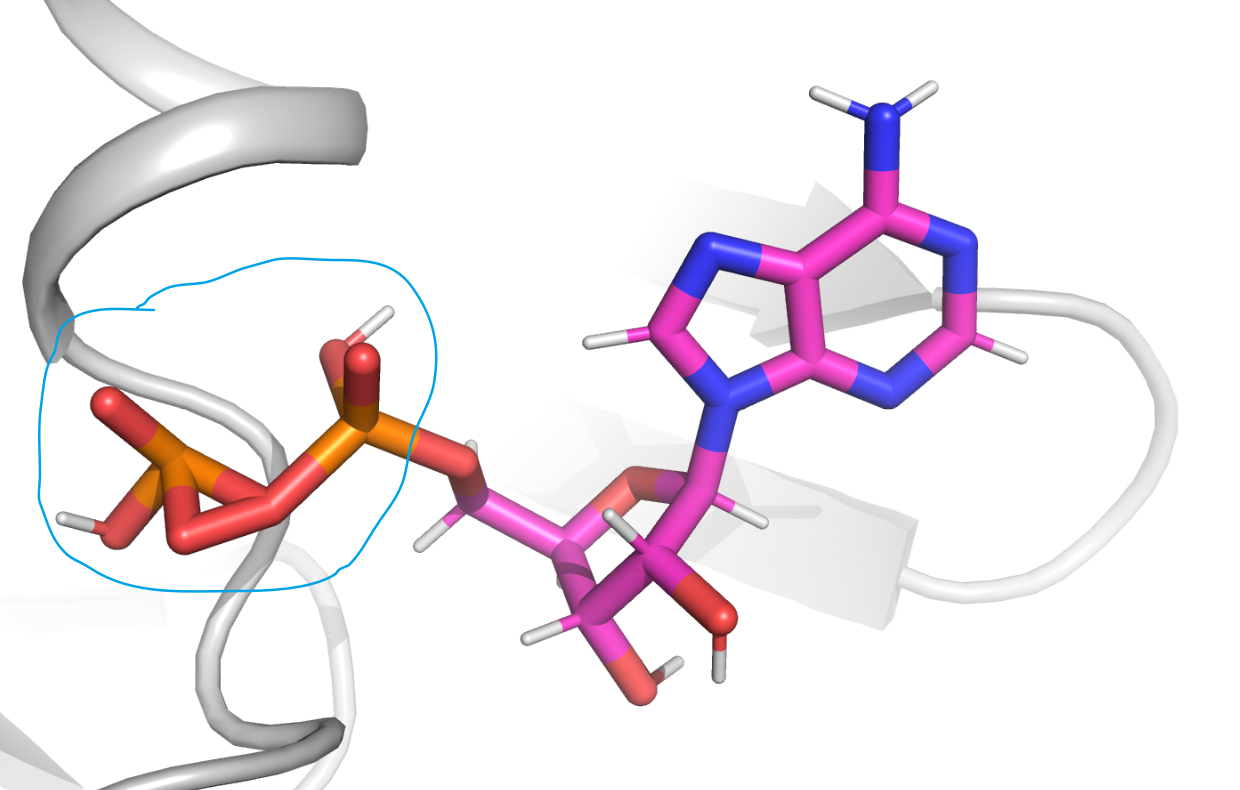
I am new for Amber. Recently, I had a fatal problem about ADP-protein complex simulation with Amber 18 GPU version, that was belonged to a blowing up type error, when the total simulated system began to heat temperature. I had also mentioned that the ADP molecule shape had been abnormal, which was shown in pic1.png file. I think this error may cause the whole simulated system blowing up. I found that the three hydrogens which were located in phosphate group, that was the possible reason about this error. Because that if the three hydrogens were missed or deleted, that error will not happen. I began to doubt that the three hydrogen atom should be added or not, despite Gaussian view don't add them automatically. So how do I handle this problem well ? Always, I employed the Gaussian view or Obabel tool to added hydrogen atom for molecule, used antechameber to caculate bcc type charge, complement the parmaters via parmchk2 program, and applyed tleap tools to finish the topology file and coordinates file building. Also, I had tried to prolong the energy minmization step to 20000 steps in the initial period, but it still doesn't work. I don't know how to solve this problem and not sure whether the three hydrogen atoms should be added. Please help me, any suggestion will be appreciated. Thanks!
Best wishes !
Ning Wang
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: adp_bcc.mol2
(image/png attachment: pic1.png)
