Date: Thu, 6 Aug 2020 19:10:35 +0800
Dear Dr. Case,
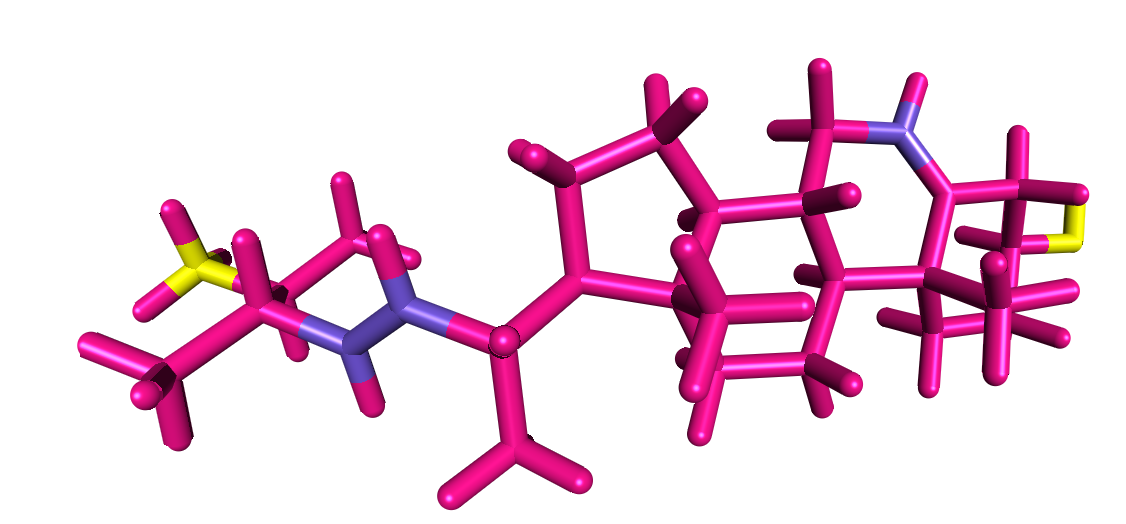
Thank you for the email. I am posting a picture I snapped in Discovery
studio visualiser
[image: image.png]
I see these three blue coloured carbons and i am not sure why they are
highlighted. are these the ones that maybe causing the error message ? But
as you said nothing looks odd about them. I have highlighted the yellow
toms that do not have an atom number assigned in the mol2 structure
"bras.mol2" I am nott sure why the software did not assign them a number.
PyRed did generate the frcmod files but there are 3 files. I am attaching
for your reference. Which one to use?
The exact command I ran when I got the error was
"antechamber -i bras.mol2 -fi mol2 -o brass.mol2 -fo mol2 -s 0 -rn LIG"
I have used the same command earlier before PyRED
I used the parmck2 for the bras.mol2. I am attaching the frcmod file it
generated. looks different from the ones generated by PyRED.
another question. which atom type file should i call for the ligand. not
sure if gaff or gaff2.
Thank you
Regards
Ayesha Fatima
On Wed, Aug 5, 2020 at 9:44 AM David A Case <david.case.rutgers.edu> wrote:
> On Tue, Aug 04, 2020, Ayesha Fatima wrote:
>
> >I would like to prepare the cholesterol molecule as ligand for md
> >simulation. previously I docked the molecule into a protein to get a
> >complex. I extracted it and prepared a charged mol2 file using the PyRED
> >server. now that want to convert to the mol2 to be used in the amber I
> >cannot generate the mol2 file. I opened the file in notepad and can see
> >that a terminal C and terminal OH atom has no atom numbers.
>
> I don't see any such atoms with no atom numbers in the "bras.mol2" file
> that
> you attached. Can you be more specific about what you found confusing
> about
> the mol2 file. I looked at the structure in Chimera, and didn't see any
> problems -- but it's a sizable molecule, and I might have missed something.
>
> > The error
> >message says "Weird atomic valence (3) for atom (ID: 34, Name: C14).
> >Possible open valence." C14 in the structure is attached to another
>
> Can you give the exact command and input files you used to get this
> message?
>
> >
> >do i need to use the antechamber after the using the PyRED server?
>
> Did pyRED provide a frcmod file to go along with the mol2 file? If so, you
> should be ready for tleap. If not, you may need to run parmchk2, but you
> should not need to run antechamber itself.
>
> ....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
- application/octet-stream attachment: frcmod.unknown
- application/octet-stream attachment: frcmod.correspondence
- application/octet-stream attachment: frcmod.known
- application/octet-stream attachment: bras.frcmod
