Date: Mon, 30 Mar 2020 11:07:58 -0400
Hello,
In the past (the way past - 2011!) we have used these parameters:
https://www.ncbi.nlm.nih.gov/pubmed/17323932
Kun Song created these leap files while in Carlos Simmerling's lab.
I attached the off and frcmod files to this email, as well as a picture.
Please make sure they are compatible with any changes to the DNA FF since
2007!
-Christina
On Mon, Mar 30, 2020 at 10:36 AM Robert Molt <rwmolt07.gmail.com> wrote:
> Good morning,
>
> I am attempting to simulate abasic DNA with a protein "bonded" to said
> DNA structure. In particular, an amino acid ends up "occupying" the
> "empty" location of the abasic DNA.
>
> In attempting to figure out the best way to construct this in Amber, I
> found the following post:
>
> http://archive.ambermd.org/201901/0211.html
>
> in which Dr. Case elaborates that one might "remove the base from A,
> replacing it with a hydrogen."
>
> I think I have a misunderstanding of how leap works for which I seek
> clarification.
>
> I have a "residue" named 48Z which is the phosphate backbone in my
> crystal structure associated with the abasic site. I think I have to
> "name" this residue something akin to "DA" or "DC" in order to tell leap
> to connect the phosphate backbone properly (it expects to connect the
> polymer). But to my understanding, the act of doing so will
> automatically tell leap to fill in the missing heavy atoms? This seems
> to be the behavior when I attempt this.
>
> --
> Dr. Robert Molt Jr.
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- ----------------------------------------------------------------- Christina Bergonzo Research Chemist Biomolecular Measurement Division, MML, NIST -----------------------------------------------------------------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: ABB.off
- application/octet-stream attachment: ABD.frcmod
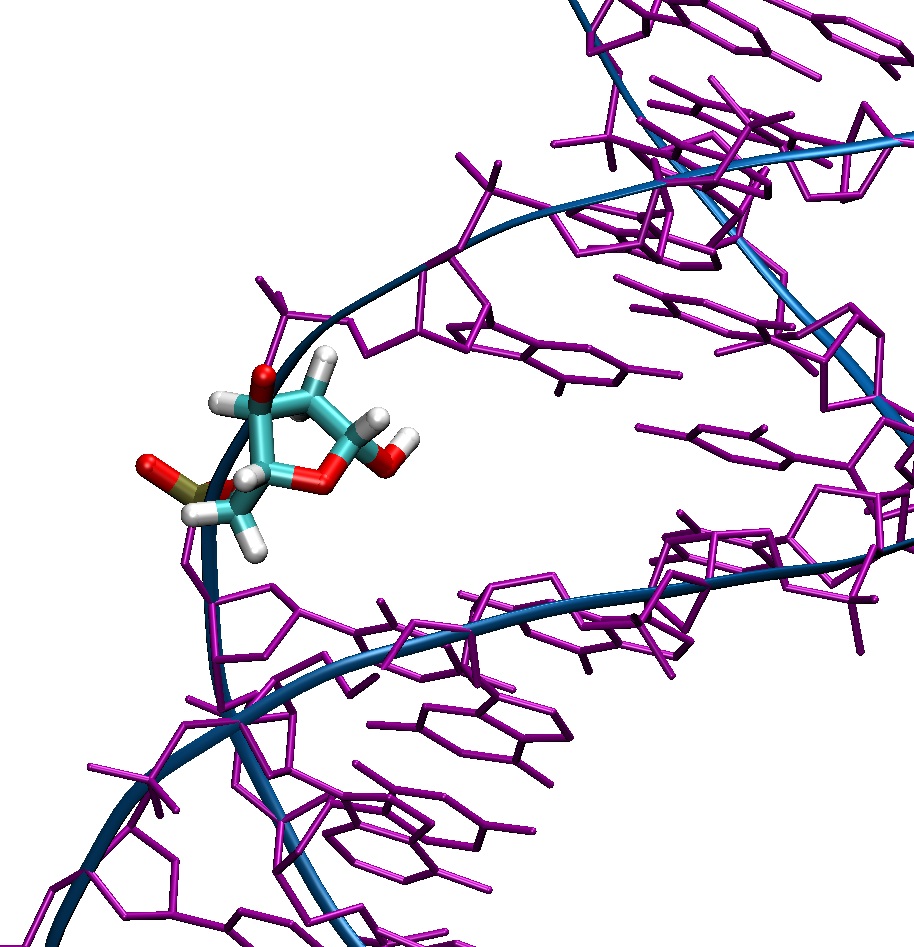
(image/jpeg attachment: abasic-site.jpg)
