Date: Tue, 17 Mar 2020 15:56:31 +0300
Dear dac,
Thanks for your answer.
I will try to go with a new example with the files to make my expression
clear: (By the way I obtained my DNA-ligand complex with autodock4.0 but
this time I will go on only ligand )
1) Here is my mol2 ligand file with resp charges :
resp.mol2 (attached)
I also change some "os" atoms to "ob" (ob: oxygen atom connec to B3)
2) I created my library with my frcmod file and gaff2:
source leaprc.gaff2
loadamberparams bor.frcmod
SUS =loadmol2 resp.mol2
saveoff SUS sus.lib
3) now I am testing sus.lib with pdb file here is resp.pdb
file: resp.pdb (attached)
source leaprc.gaff2
loadamberparams bor.frcmod
loadoff sus.lib
#once kutuphaneleri yukledik
a =loadpdb resp.pdb
saveamberparm a x.prmtop x.inpcrd
----
test:
sander -O -i min.in -o min.out -p x.prmtop -c x.inpcrd -r min1.ncrst
ambpdb -p x.prmtop -c min1.ncrst > pdb.pdb
------------
until now, everything is OK. no problem. But now, I should use ligand2.pdb
from docking result. Because this resp.pdb file was just optimized geometry
it is not inside "DNA". ligand2.pdb file is part of DNA-ligand complex.
Now, I extracted ligand2.pdb file from docking result, autodock's pdb file
format is a little bit different so, I converted this file into pdb again
in Discovery Studio Visuliaser and her is my ligand2.pdb file (extraced
from docking DNA complex):
ligand2.pdb (attached, if you look at inside, I changed residue names as
"SUS" as I described)
then I run tleap:
source leaprc.gaff2
loadamberparams bor.frcmod
loadoff sus.lib
#once kutuphaneleri yukledik
a =loadpdb ligand2.pdb
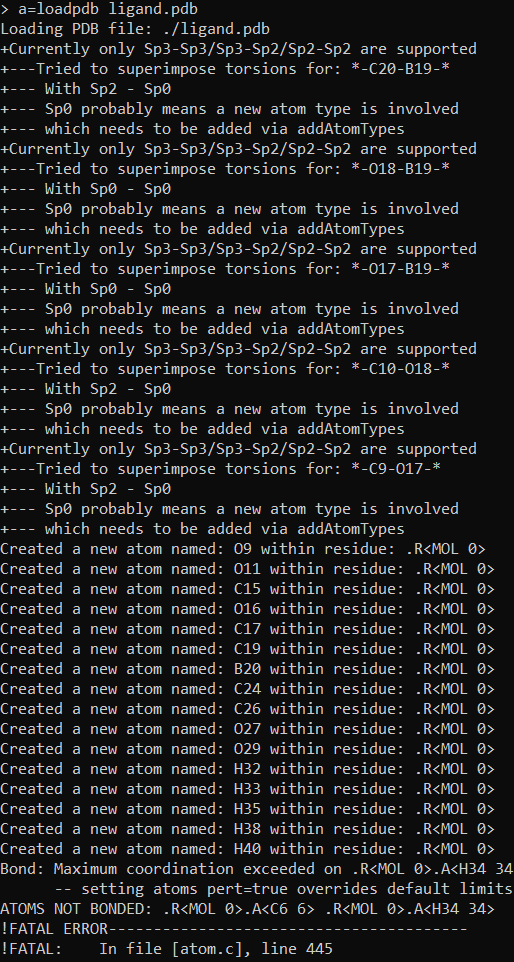
I got these warnings and tleap aborted (I shortened error message):
error.png
[image: error.png]
Created a new atom named: H40 within residue: .R<MOL 0>
Bond: Maximum coordination exceeded on .R<MOL 0>.A<H34 34>
-- setting atoms pert=true overrides default limits
ATOMS NOT BONDED: .R<MOL 0>.A<C6 6> .R<MOL 0>.A<H34 34>
!FATAL ERROR----------------------------------------
!FATAL: In file [atom.c], line 445
!FATAL: Message: bondAtomProblem found
!
!ABORTING.
Actually I can change coordinates one by one in resp.pdb file:
putting coordinates from ligand2.pdb into resp.pdb so I can obtain my
ligand2.pdb but there are 20 files as this. The order of atoms in these
files are different from each other and doing this could take too long thus
I am searching the shortest way. I did some mistakes somewhere but I
couldn't find it.
Thank you so much
David A Case <david.case.rutgers.edu>, 15 Mar 2020 Paz, 19:41 tarihinde
şunu yazdı:
> On Sat, Mar 14, 2020, Erdem Yeler wrote:
>
> >I parameterized boron and I created my *.frcmod file, for mol2 files,
> >everything was OK. I heated my molecule, calculated RMSD, minimized and
> >found very very close X-ray structure, I also calculated resp charges
> >exc.... exc... But yesterday I tried to run MMPSA for DNA-boron compund
> >complex.
>
> It's hard to reply here, but I'll try to be as clear as possible. It
> doesn't look like your problems are related to boron, but rather to
> combining a small molecule with DNA.
>
> It appears that you can run MD simualtions on the small molecule, but
> are having troubles with the DNA-boron complex.
>
> >1) I had to turn my bor.mol2 file to bor.pdb pdb file because I had to
> >use DNA.OL15 force filed too
>
> You will need to create a combined PDB file with the DNA and the small
> molecule placed in the correct place, that is docked into place. Many
> programs convert a mol2 file to pdb, including antehchamber:
>
> antechamber -fi mol2 -i resp.mol2 -fo pdb -o ligand.pdb
>
> You can create the combined pdb file either with a docking program or
> with a visualization program; or maybe(?) you already have a starting
> structure of the DNA + ligand that you can use.
> >
> > sander -O -i min.in -o min.out -p x.prmtop -c x.inpcrd -r min1.ncrst
>
> As best I can tell from your email (and maybe I missed something here),
> the "x.prtmop" file just has the small molecule. That is, I didn't see
> anything that involved tell tleap about the DNA. But you've already
> said that you were successful in running the small molecule by itself,
> which leads to my confusion.
>
> >
> >I didn't get any error message but mdin file said:"nothing" :)
>
> I don't follow you here.
>
> >
> >So I checked what happened and here is what I got :
> >
> >ambpdb -p x.prmtop -c min1.ncrst > pdb.pdb
>
> It sounds like there is indeed a problem with the x.prmtop file, but see
> above for things you might look for. If the "resp.pdb" file that you
> loaded indeed has DNA in it, be sure that there is a TER card between
> each DNA chain, and between DNA and ligand.
>
> Basically we need to know what is in the PDB file you loaded into tleap.
>
> ....dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: error.png)
- application/octet-stream attachment: resp.mol2
- application/octet-stream attachment: resp.pdb
- application/octet-stream attachment: ligand2.pdb
