Date: Thu, 12 Dec 2019 18:26:20 +0000
Hi Amber users,
I have a basic query…
I m working on a Mediator complex activator (Med25) and just out of curiosity, I minimized and equilibrated the protein in TIP3P water-box and eventually tried to see the C-alpha RMSD deviation wrt the starting structure. FYI, the starting structure is a NMR ensemble, and there were 10 conformers in the pdb file and I used each of the model in a different folder to do the minimization+ equilibration+ production (NPT).
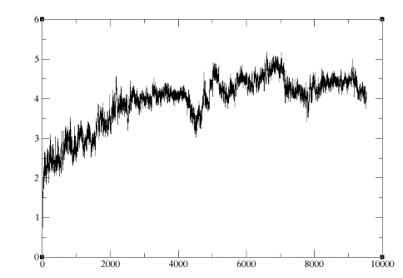
The RMSD fluctuation was in the range of 2 to 5 ANGSTROMS. This fluctuation is true of all the 10 conformers…Any thoughts on how to specifically set up the system for cosolvents based simulations or anything in particular one should be careful of while setting up this system?
Y axis RMSD, x axis no of frames.. Is it normal? The production is been run for 100 ns, using amberff14SB forcefield.
[cid:image003.png.01D5B0EF.22C16670]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 4A17B482BAE04453B0FE9F4B69D3DC47.png)
