Date: Thu, 26 Sep 2019 13:58:30 -0400
Hi,
So just looking at your reference (ref.pdb) and your input trajectory
(trajin_all.nc), it seems like you didn't strip the reference
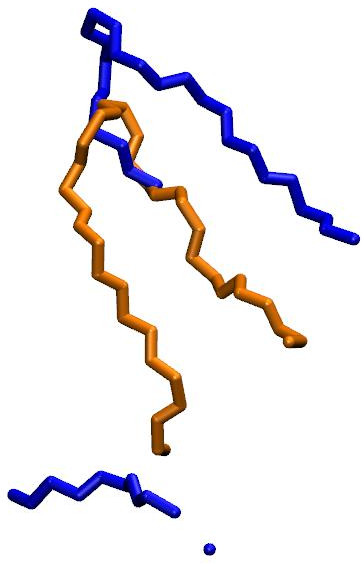
properly. I'm attaching a picture showing your reference in blue and
your trajectory in orange. Your trajectory consists of a continuous
hairpin while your reference seems to have part of a hairpin and then
some other disconnected fragments. It's not surprising that the
coordinate RMSD between these structures would be large.
It may be easier for you to just specify the reference separately,
then use 2 masks in your 'rms' command to specify the target and
reference regions. Remember, the # of atoms and the atom ordering need
to match. So it would look something like this.
# Load reference
parm refparm.parm7
reference refcoord.rst7 parmindex 0
# Load target trajectory
parm tgtparm.parm7
trajin tgtraj.nc parmindex 1
rms reference <target mask> <reference mask> out rmsd.dat
The 'select', 'atoms', 'resinfo' etc commands can be very useful for
looking at what mask expressions will actually select. Here's some
input I used to compare the residues in your two stripped topologies:
parm parm1.prmtop
parm parm2.prmtop
resinfo parm parm1.prmtop out parm1.residues
resinfo parm parm2.prmtop out parm2.residues
quit
You can then 'diff' or 'meld' the two files.
Hope this helps,
-Dan
On Mon, Sep 23, 2019 at 10:49 AM Lenka <Vellryba.seznam.cz> wrote:
>
>
> Dear Dan,
>
>
>
>
>
> I have sent you an email with Dropbox link with all the files and the
> originals. I stripped all atoms but CA,C and N.
>
>
> I only sent every 100th frame from the original trajectory due to its size
> (so there will be mismatch in number of frames).
>
>
>
>
>
> Thank you for your help.
>
>
>
>
>
> L.
>
> ---------- Původní e-mail ----------
> Od: Daniel Roe <daniel.r.roe.gmail.com>
> Komu: AMBER Mailing List <amber.ambermd.org>
> Datum: 23. 9. 2019 14:17:31
> Předmět: Re: [AMBER] cpptraj rms with different pdb
> "Hi,
>
> On Fri, Sep 20, 2019 at 3:25 PM Lenka <Vellryba.seznam.cz> wrote:
> > I have stripped all the atoms from trajectories and pdb (and associated
> > prmtops) so that there is an equal number of atoms.
> >
> > For a sanity check, I got a few frames from the trajectory and compared
> the
> > rmsd using Chimera.
> >
> > My issues is, that amber calculates the rmsd to be around 7 and Chimera
> > around 1. Since the structures are very similar, Chimera is right.
>
> As mentioned previously, given the same input and reference
> structures, cpptraj and chimera should return a best-fit RMSD within
> numerical precision.
>
> There are a few places your calculation could have gone awry - most
> notably with exactly what atoms you stripped from each structure (you
> don't say exactly what you did so no way to check if it's ok or not).
> In order for the RMSD calculation to work properly, not only the
> number of atoms needs to match but the atom ordering as well. Could
> you send me those files as well? Preferably the originals too if you
> have them. Also any other relevant information (like what atoms were
> stripped). Thanks,
>
> -Dan
>
> >
> >
> > Any idea as to what am I doing wrong?
> >
> >
> >
> >
> >
> > Thank you very much
> >
> >
> >
> >
> >
> > L.
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
> "
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: ref_and_traj.jpg)
