Date: Fri, 30 Aug 2019 15:37:52 -0600
Dear Lachele,
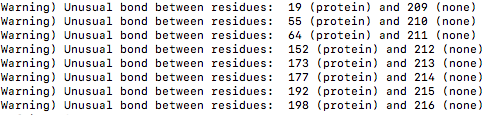
Please find attached my tleap input file. I generated the coordinate file
and topology file with tleap. When I load the topology file into VMD, I got
the following error. All the warnings are about the connections between ASN
and glycans. I checked the connections are correct, can I ignore the
warnings?
Best regards,
Rui
[image: Screen Shot 2019-08-30 at 3.37.15 PM.png]
On Mon, Aug 26, 2019 at 1:23 PM Rui Chen <rchen6.ualberta.ca> wrote:
> Dear Lachele,
>
> Got it.
>
> Thank you for your help.
>
> Best regards,
> Rui
>
> On Mon, Aug 26, 2019 at 12:31 PM Lachele Foley <lf.list.gmail.com> wrote:
>
>> 1) I'm still curious about this, but don't feel you have to spend time
>> telling me about it if you have something that works.
>>
>> 2) & 3) Yay!
>>
>> 4) The names don't matter at all to AMBER. Suffixes are irrelevant to
>> how AMBER deals with the files. It can be more convenient to you,
>> when using certain programs, such as VMD, if you name the files so
>> that the program will recognize them. My favorite suffixes are .parm7
>> and .rst7. But others are fine, too. You can simply rename them
>> however you like.
>>
>> On Sat, Aug 24, 2019 at 12:53 AM Rui Chen <rchen6.ualberta.ca> wrote:
>> >
>> > Dear Lachele,
>> >
>> > Thank you for your patience and help.
>> >
>> > 1. I used GLYCAM to generate the glycans first, however, I got severe
>> > steric clash in 2 out of 8 glycans.
>> > 2. I went to http://glycam.org/txt, checked the naming of the
>> > carbohydrate. My pdb file is correct ; ) It's very convenient to check
>> the
>> > correct naming format of specific glycans.
>> > 3. I run minimization for the glycosylated protein, it worked fine. I
>> think
>> > I am ok.
>> > 4. I generate a topology file and name if as .top, a coordinate file and
>> > name it as .crd, should I change them to .prmtop and .inpcrd,
>> respectively?
>> > If yes, can I simply rename them?
>> >
>> > Thanks again and have a nice weekend.
>> >
>> > Best regards,
>> > Rui
>> >
>> >
>> > On Fri, Aug 23, 2019 at 6:59 AM Lachele Foley <lf.list.gmail.com>
>> wrote:
>> >
>> > > 1. The easiest way is to build it using our online GUI here:
>> > > http://glycam.org/cb Another option is to enter DGlcpNAcb1-OME
>> into
>> > > the box here: http://glycam.org/txt (note the http, not https -
>> > > this is one of the smaller reasons that the current version is being
>> > > retired soon.)
>> > >
>> > > 2. I'm guessing you didn't use our online builder to add glycans to
>> > > your protein (http://glycam.org/gp)? If you were using plain leap, I
>> > > would expect some geometric issues. There are currently no formats,
>> > > that we know of or can use, that will encode complex geometric
>> > > information such as one finds with carbohydrates. We are currently
>> > > working on encoding formats that will allow a system such as leap to
>> > > understand such complexities. For now, these need to be set manually
>> > > or by heuristics built in to software like our Glycoprotein Builder
>> > > GUI or CHARMM's.
>> > >
>> > > The force field is independent of the initial geometry. If an initial
>> > > geometry isn't terrible, a force field can be used to minimize the
>> > > system and improve the geometry. But, if the initial geometry is
>> > > really bad, a force field (generally) will not correct the issue.
>> > > This isn't a problem; to the contrary, it is desirable behavior. If,
>> > > in nature, you were to somehow place two atoms so close to each other
>> > > that their cores are very close to each other, the system would be
>> > > unstable. The force field only tries, incompletely, to mimic the
>> > > behavior of nature. The CHARMM gui is doing some geometric
>> > > manipulations for you in the background. With leap, you simply must
>> > > do them yourself.
>> > >
>> > > If leap built you a prmtop and inpcrd, you should be ok. But, run a
>> > > little simulation to be sure, of course.
>> > >
>> > > On Thu, Aug 22, 2019 at 10:21 PM Rui Chen <rchen6.ualberta.ca> wrote:
>> > > >
>> > > > Dear Lachele,
>> > > >
>> > > > 1. Where can I download the library for GlcpNAcb1-OME?
>> > > >
>> > > > 2. I used CHARMM GUI to generate the glycans, now I am running
>> Amber, in
>> > > > the leap input file, I sourced the GLYCAM force field, even though
>> it
>> > > > worked, I think that's not correct. The reason I use CHARMM GUI to
>> > > generate
>> > > > the glycans not GLYCAM is that I tried to use GLYCAM to generate the
>> > > > glycans, I got severe steric clash, CHARMM GUI worked fine and
>> easier. Do
>> > > > you have any suggestion about the force field?
>> > > >
>> > > > Best regards,
>> > > > Rui
>> > > >
>> > > >
>> > > >
>> > > >
>> > > > On Thu, Aug 22, 2019 at 9:45 AM Rui Chen <rchen6.ualberta.ca>
>> wrote:
>> > > >
>> > > > > Thank you for your help. Where can I download the library
>> > > > > for GlcpNAcb1-OME? I couldn't find it.
>> > > > >
>> > > > >
>> > > > >
>> > > > > On Thu, Aug 22, 2019 at 8:24 AM Lachele Foley <lf.list.gmail.com>
>> > > wrote:
>> > > > >
>> > > > >> 1. Probably. The only way to be certain is to download a
>> > > > >> GlcpNAcb1-OME from our site and open them both up in
>> VMD/Chimera/etc
>> > > > >> and compare the atoms to each other and assign names based on
>> that
>> > > > >> comparison. I think you also need to change C to C2N and O to
>> O2N.
>> > > > >>
>> > > > >> 2. Yes.
>> > > > >>
>> > > > >> 3. If all goes according to plan, yes. If this is not what
>> happens,
>> > > > >> then something went wrong. The NAc group in GlcNAc is often a
>> little
>> > > > >> bent in PDB files.
>> > > > >>
>> > > > >> On Wed, Aug 21, 2019 at 10:18 PM Rui Chen <rchen6.ualberta.ca>
>> wrote:
>> > > > >> >
>> > > > >> > No problem.
>> > > > >> > Just want to double check:
>> > > > >> > 1. I renamed N to N2, CT to CME, that's correct.
>> > > > >> > 2. In order to avoid the problem of hydrogen naming, I delete
>> all
>> > > the
>> > > > >> > hydrogens, since leap will generate the hydrogens for me.
>> > > > >> > 3. I don't need to worry about the improper torsions,
>> minimization
>> > > will
>> > > > >> > solve the problem.
>> > > > >> > Thank you.
>> > > > >> >
>> > > > >> > On Wed, Aug 21, 2019 at 7:36 PM Lachele Foley <
>> lf.list.gmail.com>
>> > > > >> wrote:
>> > > > >> >
>> > > > >> > > We are currently working on the inclusion of glycan
>> processing in
>> > > > >> > > PDB4AMBER. At the moment, its capabilities in that regard
>> are a
>> > > > >> > > little limited. Our biggest holdup right now is getting some
>> > > graph
>> > > > >> > > pattern matching to work. The latter will help make the atom
>> > > renaming
>> > > > >> > > robust even if some atoms are missing, etc. If you know
>> anyone
>> > > who
>> > > > >> > > knows a lot about graph comparisons in the boost libraries,
>> send
>> > > them
>> > > > >> > > to us! :-)
>> > > > >> > >
>> > > > >> > > On Wed, Aug 21, 2019 at 7:02 PM Rui Chen <rchen6.ualberta.ca
>> >
>> > > wrote:
>> > > > >> > > >
>> > > > >> > > > Dear Lachele,
>> > > > >> > > >
>> > > > >> > > > I modified the PDB file according to the naming format you
>> sent
>> > > to
>> > > > >> me,
>> > > > >> > > > there is no specific type for N, I guess it should be N2,
>> no
>> > > > >> specific
>> > > > >> > > type
>> > > > >> > > > for CT, I guess it should be CME. I changed them. Now in my
>> > > leap.log
>> > > > >> > > file,
>> > > > >> > > > I only got warning about improper torsion angles.
>> > > > >> > > >
>> > > > >> > > > It seems like it's better to delete all the H before
>> running
>> > > leap.
>> > > > >> Since
>> > > > >> > > > PDB4AMBER will generate H, I removed all the H by VMD and
>> then
>> > > leap.
>> > > > >> > > >
>> > > > >> > > > I not not sure what's the exact purpose of PDB4AMBER,
>> because
>> > > for
>> > > > >> me I
>> > > > >> > > > still need to manually modify everything.
>> > > > >> > > > [image: Screen Shot 2019-08-21 at 4.29.20 PM.png]
>> > > > >> > > >
>> > > > >> > > > Thanks a lot for your help, very helpful.
>> > > > >> > > >
>> > > > >> > > > Best regards,
>> > > > >> > > > Rui
>> > > > >> > > >
>> > > > >> > > > On Tue, Aug 20, 2019 at 11:32 PM Lachele Foley <
>> > > lf.list.gmail.com>
>> > > > >> > > wrote:
>> > > > >> > > >
>> > > > >> > > > > > Thank you for the quick reply. Please let me know
>> where can
>> > > I
>> > > > >> find
>> > > > >> > > the
>> > > > >> > > > > > naming templates.
>> > > > >> > > > >
>> > > > >> > > > > 0YB is in
>> > > > >> > > > >
>> > > > >> > >
>> > > > >>
>> > >
>> http://glycam.org/docs/forcefield/wp-content/uploads/sites/6/2014/03/GLYCAM_06j-1.prep
>> > > > >> > > > > Search the page to find the 0YB entry.
>> > > > >> > > > >
>> > > > >> > > > > NLN for mid-chain positions is in
>> > > > >> > > > >
>> > > > >> > > > >
>> > > > >> > >
>> > > > >>
>> > >
>> http://glycam.org/docs/forcefield/wp-content/uploads/sites/6/2014/02/GLYCAM_amino_06j_12SB.lib
>> > > > >> > > > > Again, use a search to find that entry. I think your
>> atoms
>> > > are
>> > > > >> named
>> > > > >> > > > > right in NLN, but didn't do a careful check. Leap will
>> tell
>> > > you
>> > > > >> all
>> > > > >> > > > > about it if not.
>> > > > >> > > > >
>> > > > >> > > > > Both file formats are a little strange, but should be
>> mostly
>> > > > >> readable.
>> > > > >> > > > > More info on them is here:
>> http://ambermd.org/formats.html
>> > > > >> > > > > The first is a prep file and the second is an off file.
>> > > > >> > > > >
>> > > > >> > > > > > 1. By the way, PDB4AMBER generated the TER cards after
>> each
>> > > > >> NLN, I
>> > > > >> > > can
>> > > > >> > > > > > manually delete them.
>> > > > >> > > > >
>> > > > >> > > > > Ah. Leap will turn that residue and the next into their
>> > > > >> zwitterionic
>> > > > >> > > > > forms if there is a TER card. Best to delete them. You
>> > > might be
>> > > > >> > > > > able to override that with some sort of leap instruction.
>> > > I'm not
>> > > > >> > > > > sure. Let me know if you have trouble after deleting
>> them.
>> > > > >> > > > >
>> > > > >> > > > > > 2. NLN are 0YB are the correct naming. But as you can
>> see
>> > > in my
>> > > > >> leap
>> > > > >> > > > > input
>> > > > >> > > > > > file, I already source the force field specific for
>> glycans.
>> > > > >> Now the
>> > > > >> > > only
>> > > > >> > > > > > problem is the glycan.
>> > > > >> > > > > > [image: Screen Shot 2019-08-20 at 10.23.18 PM.png]
>> > > > >> > > > >
>> > > > >> > > > > The 0YB *residue* is named correctly. It's the atoms
>> that
>> > > aren't.
>> > > > >> > > > > For example, there are no atoms in the template named N,
>> C,
>> > > O, CT,
>> > > > >> > > > > etc. I think NLN's atoms are ok.
>> > > > >> > > > >
>> > > > >> > > > > This is also the issue with the hydrogens. They all
>> have to
>> > > be
>> > > > >> named
>> > > > >> > > > > correctly or leap fails.
>> > > > >> > > > >
>> > > > >> > > > > > 3. I used PDB2PQR to predict the protonation states of
>> the
>> > > > >> protein,
>> > > > >> > > > > PDB2PQR
>> > > > >> > > > > > generates the hydrogens. I manually deleted all the
>> > > hydrogens.
>> > > > >> Does
>> > > > >> > > it
>> > > > >> > > > > > influence the protonation state of the protein?
>> > > > >> > > > >
>> > > > >> > > > > I suppose you're talking about histidine residues? So
>> long as
>> > > > >> you've
>> > > > >> > > > > named them for the protonation state you want (HID, HIE
>> or
>> > > HIP),
>> > > > >> leap
>> > > > >> > > > > should add in the correct number of hydrogens and assign
>> > > > >> reasonable
>> > > > >> > > > > partial charges to them. I didn't find any instances of
>> > > "HIS" in
>> > > > >> your
>> > > > >> > > > > file, but I did find HID and HIE, so I think you're ok.
>> > > > >> > > > >
>> > > > >> > > > >
>> > > > >> > > > >
>> > > > >> > > > > > On Tue, Aug 20, 2019 at 10:07 PM Lachele Foley <
>> > > > >> lf.list.gmail.com>
>> > > > >> > > > > wrote:
>> > > > >> > > > > >
>> > > > >> > > > > > > Hi!
>> > > > >> > > > > > >
>> > > > >> > > > > > > You shouldn't use TER cards after the NLNs unless
>> they are
>> > > > >> > > actually at
>> > > > >> > > > > > > the end of an amino acid chain. They are amino acid
>> > > > >> (modified),
>> > > > >> > > and
>> > > > >> > > > > > > don't follow the glycan rules.
>> > > > >> > > > > > >
>> > > > >> > > > > > > Removing hydrogens isn't necessary, but their names
>> need
>> > > to
>> > > > >> exactly
>> > > > >> > > > > > > match the templates in the force fields. Folks
>> usually
>> > > > >> prefer to
>> > > > >> > > just
>> > > > >> > > > > > > remove them rather than rename them all. If any of
>> their
>> > > > >> positions
>> > > > >> > > > > > > are critical, leave those in and make sure the names
>> are
>> > > > >> right.
>> > > > >> > > > > > > Otherwise, it's easier to delete them. Or rename
>> them
>> > > all if
>> > > > >> you
>> > > > >> > > > > > > prefer to do that.
>> > > > >> > > > > > >
>> > > > >> > > > > > > Your NLN and 0YB atom names need to exactly match the
>> > > > >> templates as
>> > > > >> > > > > > > well. The same hydrogen-naming logic applies, but
>> you
>> > > must
>> > > > >> ensure
>> > > > >> > > > > > > that the heavy-atom names match.
>> > > > >> > > > > > >
>> > > > >> > > > > > > Do you know how to find the lib and prep files where
>> the
>> > > > >> templates
>> > > > >> > > are
>> > > > >> > > > > > > (for naming)?
>> > > > >> > > > > > >
>> > > > >> > > > > > > On Tue, Aug 20, 2019 at 11:49 PM Rui Chen <
>> > > rchen6.ualberta.ca
>> > > > >> >
>> > > > >> > > wrote:
>> > > > >> > > > > > > >
>> > > > >> > > > > > > > By the way, should I remove all the hydrogens
>> before
>> > > using
>> > > > >> > > PDB4AMBER
>> > > > >> > > > > and
>> > > > >> > > > > > > > why? Thank you.
>> > > > >> > > > > > > >
>> > > > >> > > > > > > > On Tue, Aug 20, 2019 at 9:43 PM Rui Chen <
>> > > > >> rchen6.ualberta.ca>
>> > > > >> > > wrote:
>> > > > >> > > > > > > >
>> > > > >> > > > > > > > > Hello,
>> > > > >> > > > > > > > >
>> > > > >> > > > > > > > > I am preparing a PDB file for running LEaP.
>> Since I
>> > > add
>> > > > >> glycans
>> > > > >> > > > > using
>> > > > >> > > > > > > > > CHARMM GUI, I rename the glycans to NLN
>> (N-linkages to
>> > > > >> ASN)
>> > > > >> > > > > according
>> > > > >> > > > > > > to
>> > > > >> > > > > > > > > AMBER manual (LEaP). However, I got error when I
>> run
>> > > > >> leap. I
>> > > > >> > > > > changed
>> > > > >> > > > > > > the
>> > > > >> > > > > > > > > TER cards, but it still didn't work. I also
>> checked
>> > > the
>> > > > >> > > connections
>> > > > >> > > > > > > between
>> > > > >> > > > > > > > > the carbohydrates and the ASN amino acids using
>> VMD, I
>> > > > >> couldn't
>> > > > >> > > > > find
>> > > > >> > > > > > > > > anything wrong. The leap input file, log file,
>> pdb
>> > > file
>> > > > >> > > (mentioned
>> > > > >> > > > > in
>> > > > >> > > > > > > leap
>> > > > >> > > > > > > > > input file) are attached.
>> > > > >> > > > > > > > >
>> > > > >> > > > > > > > > Could you please give me some clue about how to
>> solve
>> > > the
>> > > > >> > > problem?
>> > > > >> > > > > > > Looking
>> > > > >> > > > > > > > > forward to hearing from you.
>> > > > >> > > > > > > > >
>> > > > >> > > > > > > > > Sincerely,
>> > > > >> > > > > > > > > Rui
>> > > > >> > > > > > > > >
>> > > > >> > > > > > > > _______________________________________________
>> > > > >> > > > > > > > AMBER mailing list
>> > > > >> > > > > > > > AMBER.ambermd.org
>> > > > >> > > > > > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >> > > > > > >
>> > > > >> > > > > > >
>> > > > >> > > > > > >
>> > > > >> > > > > > > --
>> > > > >> > > > > > > :-) Lachele
>> > > > >> > > > > > > Lachele Foley
>> > > > >> > > > > > > CCRC/UGA
>> > > > >> > > > > > > Athens, GA USA
>> > > > >> > > > > > >
>> > > > >> > > > > > > _______________________________________________
>> > > > >> > > > > > > AMBER mailing list
>> > > > >> > > > > > > AMBER.ambermd.org
>> > > > >> > > > > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >> > > > > > >
>> > > > >> > > > > > _______________________________________________
>> > > > >> > > > > > AMBER mailing list
>> > > > >> > > > > > AMBER.ambermd.org
>> > > > >> > > > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >> > > > >
>> > > > >> > > > >
>> > > > >> > > > >
>> > > > >> > > > > --
>> > > > >> > > > > :-) Lachele
>> > > > >> > > > > Lachele Foley
>> > > > >> > > > > CCRC/UGA
>> > > > >> > > > > Athens, GA USA
>> > > > >> > > > >
>> > > > >> > > > > _______________________________________________
>> > > > >> > > > > AMBER mailing list
>> > > > >> > > > > AMBER.ambermd.org
>> > > > >> > > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >> > > > >
>> > > > >> > > > _______________________________________________
>> > > > >> > > > AMBER mailing list
>> > > > >> > > > AMBER.ambermd.org
>> > > > >> > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >> > >
>> > > > >> > >
>> > > > >> > >
>> > > > >> > > --
>> > > > >> > > :-) Lachele
>> > > > >> > > Lachele Foley
>> > > > >> > > CCRC/UGA
>> > > > >> > > Athens, GA USA
>> > > > >> > >
>> > > > >> > > _______________________________________________
>> > > > >> > > AMBER mailing list
>> > > > >> > > AMBER.ambermd.org
>> > > > >> > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >> > >
>> > > > >> > _______________________________________________
>> > > > >> > AMBER mailing list
>> > > > >> > AMBER.ambermd.org
>> > > > >> > http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >>
>> > > > >>
>> > > > >>
>> > > > >> --
>> > > > >> :-) Lachele
>> > > > >> Lachele Foley
>> > > > >> CCRC/UGA
>> > > > >> Athens, GA USA
>> > > > >>
>> > > > >> _______________________________________________
>> > > > >> AMBER mailing list
>> > > > >> AMBER.ambermd.org
>> > > > >> http://lists.ambermd.org/mailman/listinfo/amber
>> > > > >>
>> > > > >
>> > > > _______________________________________________
>> > > > AMBER mailing list
>> > > > AMBER.ambermd.org
>> > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > >
>> > >
>> > >
>> > > --
>> > > :-) Lachele
>> > > Lachele Foley
>> > > CCRC/UGA
>> > > Athens, GA USA
>> > >
>> > > _______________________________________________
>> > > AMBER mailing list
>> > > AMBER.ambermd.org
>> > > http://lists.ambermd.org/mailman/listinfo/amber
>> > >
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org
>> > http://lists.ambermd.org/mailman/listinfo/amber
>>
>>
>>
>> --
>> :-) Lachele
>> Lachele Foley
>> CCRC/UGA
>> Athens, GA USA
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screen_Shot_2019-08-30_at_3.37.15_PM.png)
- application/octet-stream attachment: 1I8LA.in
