Date: Thu, 24 Jan 2019 15:37:26 +0000
Dear Amber Users,
I am trying to run constant pH MD (CpHMD) with AMBER, in combination with accelerated MD (aMD). I am using explicit water.
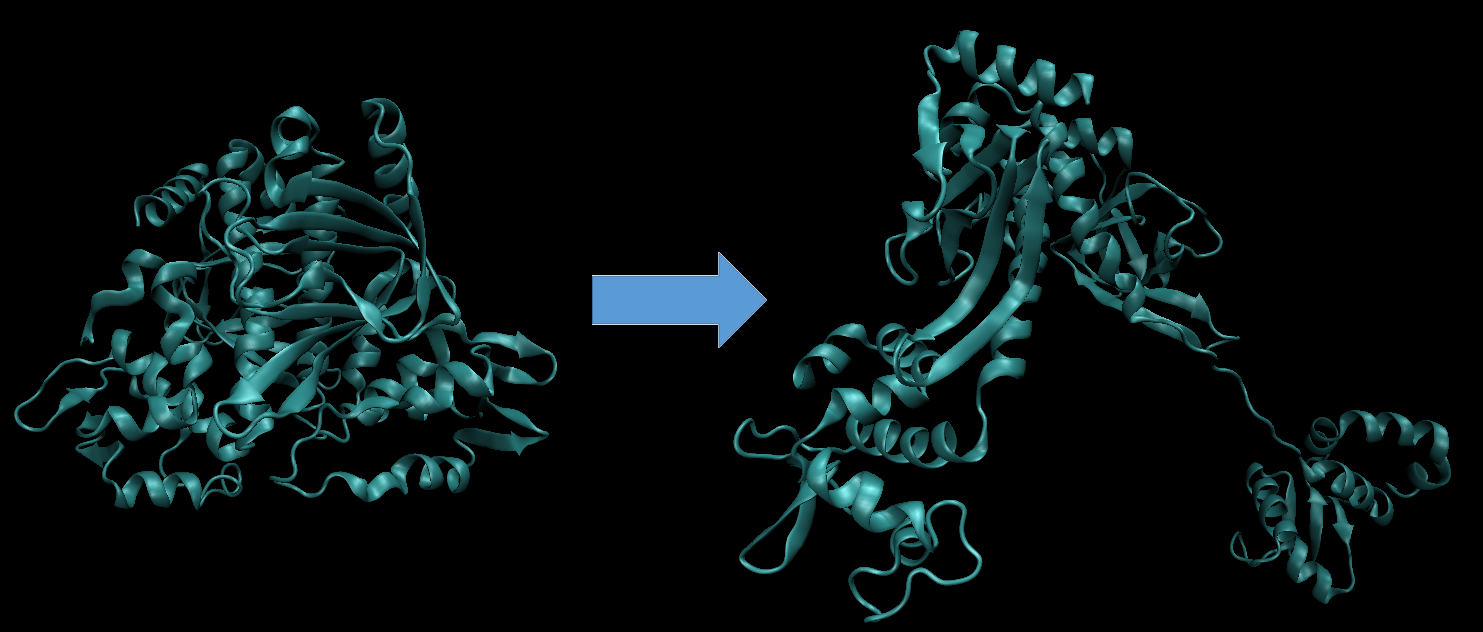
However, when I use these settings the protein unfolds, as if it is being "pulled apart". I have attached a figure from the beginning of the MD simulation and from when the protein starts to unfold. This simulation included 10 ns of CpHMD, followed by 10 ns of CpHMD in combination with aMD. The unfolding starts almost immediately after aMD is started.
I have restarted the simulation twice but the problem remains.
If I use CpHMD without trying to combine it with aMD, it looks ok during the 70 ns I have run it for. (However, it is clearly more dynamic than when running normal MD.)
Has anyone else tried this? I can only find examples of implicit MD where CpHMD and aMD are combined. Is it possible, or do I need to use implicit water instead of explicit in case I want to combine CpHMD with aMD?
I have also attached the settings I use for the production run.
Best regards,
Cecilia
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: cphmd_amd_combined.png)
- application/octet-stream attachment: 6_production_aMD.in
