Date: Fri, 14 Sep 2018 11:13:16 -0400
All,
I am trying to run an SMD simulation and am noticing some odd behavior in
the output. For context, I have two proteins and am trying to interrupt the
interface. In order to have control of what direction I pull from, I've
introduced a pseduoatom into the simulation. After locking the coordinates
of the pseudoatom and protein 1, I decrease the distance from the CoM of
protein 2 and the pseudoatom. Once I generate the simulation, I calculated
the distance from the CoM of protein 2 and the pseudoatom, and as expected
the distance does decrease. However, I notice large jumps along the
reaction coordinate, sometimes jumps increasing and sometimes jumps
decreasing the distance between the two. When visualizing the trajectory it
genuinely looks massive changes in the actual coordinates of the protein.
I had guessed that accessing the trajectory while it was still being
written to was introducing these errors (ThreeAccess image) as every time I
trajin'd the trajectory it would introduce a jump. As such, I started a new
simulation that I didn't access expecting it to fix the issue, but I'm
getting the same error. Does anyone have any thoughts as to what may be
going on?
Many Thanks,
Cory Ayres
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
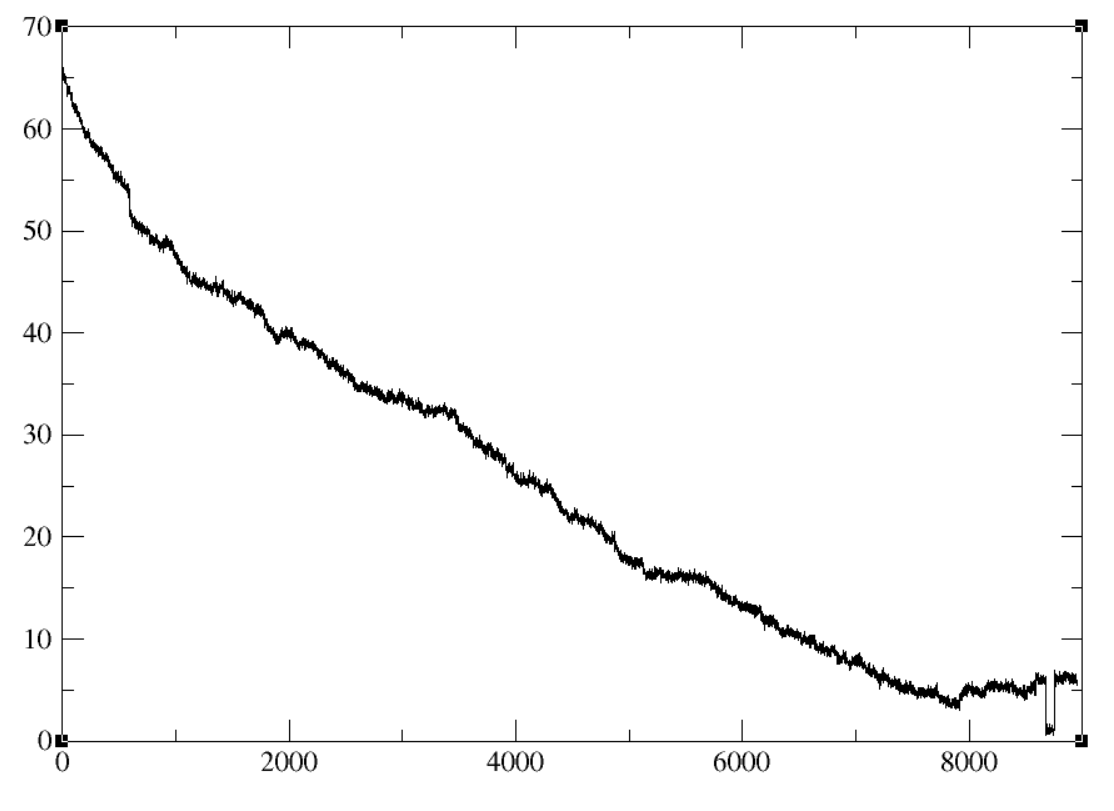
(image/png attachment: NewSimulation_NoAccess.PNG)
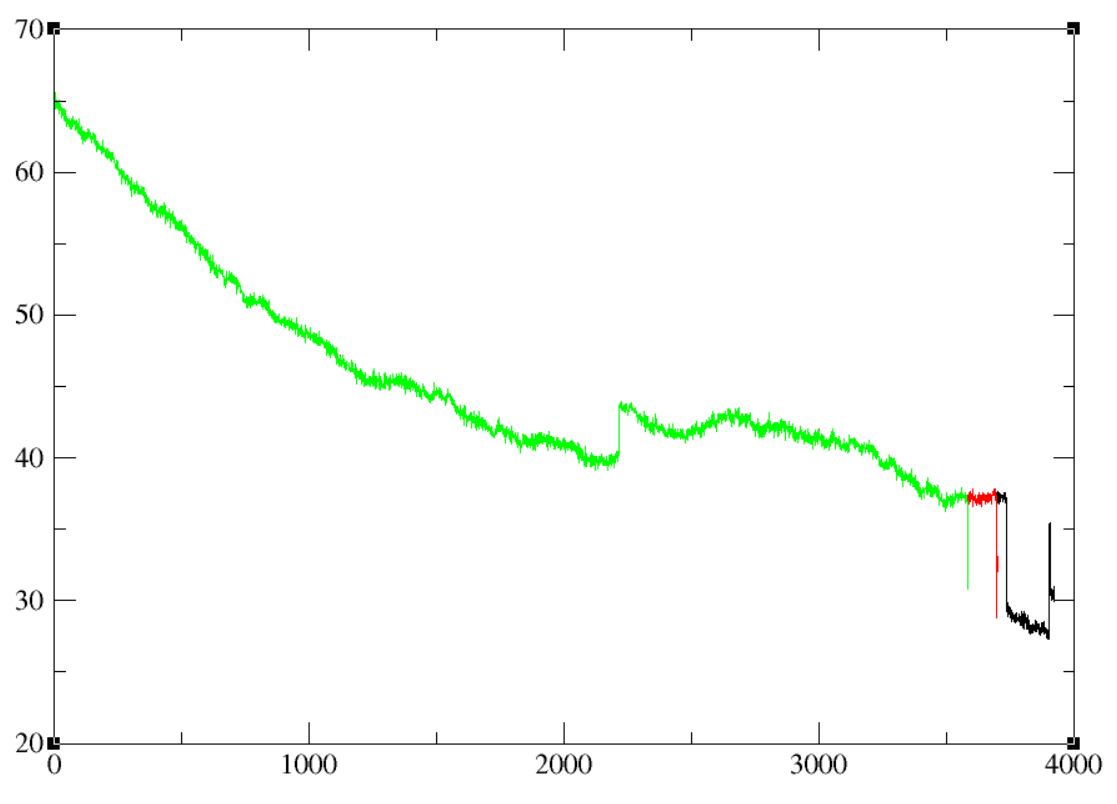
(image/png attachment: OriginalSimulation_Three_Access.PNG)
