Date: Fri, 14 Sep 2018 21:25:56 +0800 (CST)
Dear AMBER,
Firstly, thanks for your detailed answer to my last question.
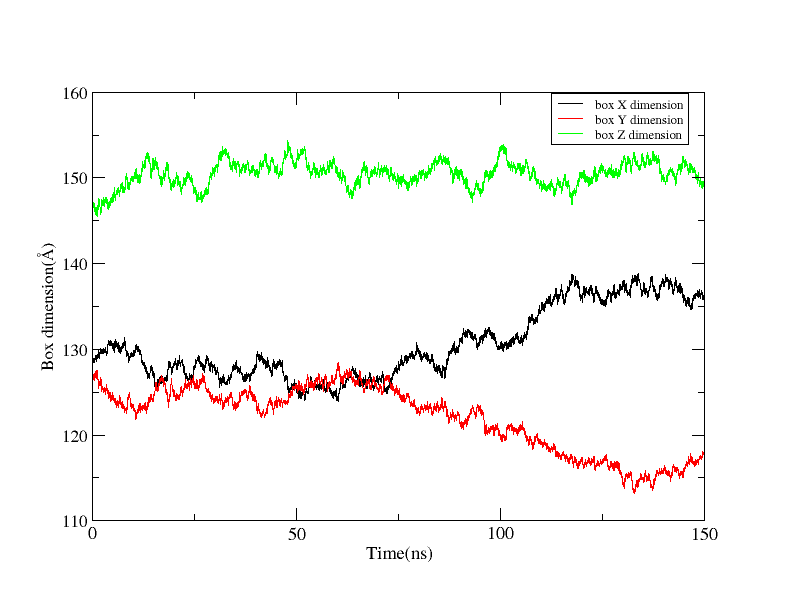
I simulate large system (~240 thousand atoms) of membrane protein dimer embedded inside a POPC membrane. The system has ran 150ns completed through three consecutive runs. But the dimension of the periodic box in the X,Y direction changes too much.
the command in "prod.in" is:
50ns
&cntrl
imin=0, ntx=5, irest=1,
ntc=2, ntf=2, tol=0.0000001,
nstlim=25000000, ntt=3, gamma_ln=2.0,
temp0=303.0,
iwrap=1,
ntpr=5000, ntwr=5000, ntwx=5000,
dt=0.002, ig=-1,
ntb=2,ntp=2,cut=10.0,ioutfm=1,
/
Is this normal? Or is there any problem with the pressure of my system?
Thank you in advance.
Zhenmei Gao
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: z.png)
