Date: Fri, 7 Sep 2018 02:14:59 +0000
Dear sir,
Thanks for your answer! By the way, I have another question: I want to calculate the interaction energy between my protein which is a channel and the transport substrate molecules and then do the energy decomposition of the interaction energy into other energy terms such as ele and vdw to find the key residues that contribute a lot. I have found the keywords "energy" and "lie" in cpptraj, but I don't know which one could get what I want. And I have researched some papers which did the similar analysis by using energy rerun with gromacs, does amber have the similar methods?
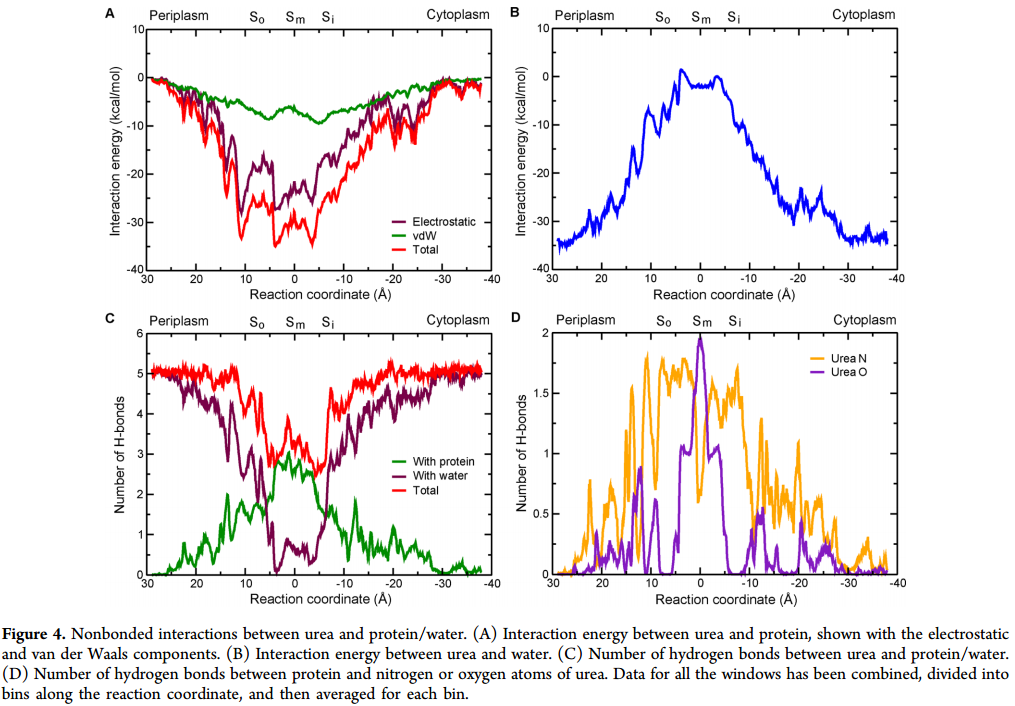
ps: Here I attached a picture from a paper (https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.6b00602), and the Figure4 is what I want!
All the best,
Meng Wu
------------------------------
On Thu, 6 Sep 2018 14:59:39 David A Case <david.case.rutgers.edu> wrote:
> On Thu, Sep 06, 2018, Meng Wu wrote:
>
> I am going to analyze the energy terms during the MD process, here
> I have two quick questions, 1) What's the energy units in mden file
> and mdout_analyzer.mdout_analyzer.pypy outcomes? Is kcal/mol or
> kJ/mol ? 2) What's the difference between the outcomes of mden file
> and mdout_analyzer.py? Are they the same or not?
Energies are printed to mdout every ntpr steps, whereas energies and printed
to the mden file every ntwe steps. So you may have different amounts of
information in the mdout vs. the mden file.
All Amber energies are in kcal/mol.
....dac
------------------------------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Figure4.png)
