Date: Fri, 6 Jul 2018 13:32:28 +0000 (UTC)
Thank you for the script and also for the note on the ffs.
Regarding the ionic strength:
The box has indeed shrank from 485952.034 A^3 to 451488.3734 A^3 in a simulation, in which 19 Na+ ions are added to neutralise the negative charge of the DNA and packed the solvent with 0.75 closeness. I modified your script to take the volume reported by in the md.out so I could use it for a truncated octahedron box.
10mM NaCl: 2.8 molecules of NaCl + 19 Na+ ions to neutralise the DNA > 2.8 anions + 21.8 cations
100mM NaCl: 28.1 molecules of NaCl + 19 Na+ ions to neutralise the DNA > 28.1 antions + 47.1 cations
500mM NaCl: 140.7 molecules of NaCl + 19 Na+ ions to neutralise the DNA > 140.7 anions + 159.7 cations
1M NaCl: 280.8 molecules of NaCl + 19 Na+ ions to neutralise the DNA > 280.8 anions + 299.9 cations
I also tried SLTCAP webcalculator, although there I could only input a cubic box with comparable volume instead of the truncated octahedron. That calculation does indeed gives lower number of ions that need to be added:
10 mM NaCl: Your system requires 0.32 anions and 19.32 cations.
100 mM NaCl: Your system requires 17.21 anions and 36.21 cations.
500 mM NaCl: Your system requires 115.69 anions and 134.69 cations.
1000 mM NaCl: Your system requires 240.33 anions and 259.33 cations.
mass: 6KDa
net charge of solute: -19
cubic box length 7.5 nm
number of water molecules ca. 14400
Distance from protein to box edge (nm): 4
Longest axis of protein (nm): 7
Still the question is where the ions should be put during the building of the model: according to electrostatic potential with addions(2) or randomly with addIonsRand? Monovalent cations were shown to distribute themselves in the grooves close to the bases instead of the phosphate groups according to the work of Richard Lavery.
Best,Kris
On Friday, July 6, 2018, 1:44:45 AM GMT+2, Thomas Cheatham <tec3.utah.edu> wrote:
> I would like to run simulations on ssDNA under 10 mM, 100 mM, 500 mM and 1 M NaCl concentrations by adding the necessary number of Na+ and Cl- ions. Is there any way of specifying this so that the number of ions are calculated automatically? Something similar exists in GROMACS:
> for 100 mM:
> gmxgenion -s ions.tpr -o protein_solv_ions.gro -p topol.top -pname NA -nname CL -neutral-conc 0.1
>
> I checked addIons, addIons2 and addIonsRand, I searched the mailing list, but I do not see this feature.
No, you have to figure out how much you need by hand noting that standard
LEaP built water boxes will shrink a little unless one is clever in
building it (by altering closeness in the solvate commands)... I have an
old Perl script that I use (appended) to figure out how many ions I need.
Also note that many of the AMBER force fields do not necessarily well
represent single strands (either metastable duplex like structure on short
timescales like ~500ns-1 microsecond or over-compacted on longer
timescales). This was a focus effort for force field improvement with the
DE Shaw nucleic acid force field(s).
Proc Natl Acad Sci U S A. 2018 Feb 13;115(7):E1346-E1355.
doi: 10.1073/pnas.1713027115.
"RNA force field with accuracy comparable to state-of-the-art protein
force fields." Tan D, Piana S, Dirks RM, Shaw DE.
Also, for assessing needed DNA salt concentration, there is a recent
article by Smith and co-workers:
https://pubs.acs.org/doi/10.1021/acs.jctc.7b01254
--tec3
Perl:
% more ~/perl/molarity.perl
#!/usr/sbin/perl
$molar = $ARGV[0];
$box_x = $ARGV[1];
$box_y = $ARGV[2];
$box_z = $ARGV[3];
$box_a = $ARGV[4];
$box_b = $ARGV[5];
$box_g = $ARGV[6];
if ($box_x == 0) {
printf("Usage: molarity box-x box-y box-z alpha beta gamma\n");
die;
}
if ($box_y == 0) {
$box_y = $box_x;
}
if ($box_z == 0) {
$box_z = $box_x;
}
if ($box_a == 0) {
$box_a = 90.0;
}
if ($box_b == 0) {
$box_b = $box_a;
}
if ($box_g == 0) {
$box_g = $box_a;
}
$torad = 2 * 3.141592654 / 360.0;
$rad_a = $box_a * $torad;
$rad_b = $box_b * $torad;
$rad_g = $box_g * $torad;
$angles = 1 - cos($rad_a)*cos($rad_a) - cos($rad_b)*cos($rad_b) -
cos($rad_g)*cos($rad_g);
$angles += 2 * cos($rad_a)*cos($rad_b)*cos($rad_g);
$angles = sqrt($angles);
$volume = $box_x * $box_y * $box_z * $angles;
$molecules = 6.022 * $volume * $molar / 10000;
printf(" MOLARITY = %8.3f\n", $molar);
printf(" Box size = %8.3f %8.3f %8.3f %8.3f %8.3f %8.3f\n", $box_x,
$box_y, $box_z, $box_a, $box_b, $box_g);
printf(" Volume = %8.3f\n", $volume);
printf("\n %8.3f molecules are necessary to make a molarity of %6.2f
M\n\n", $molecules, $molar);
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
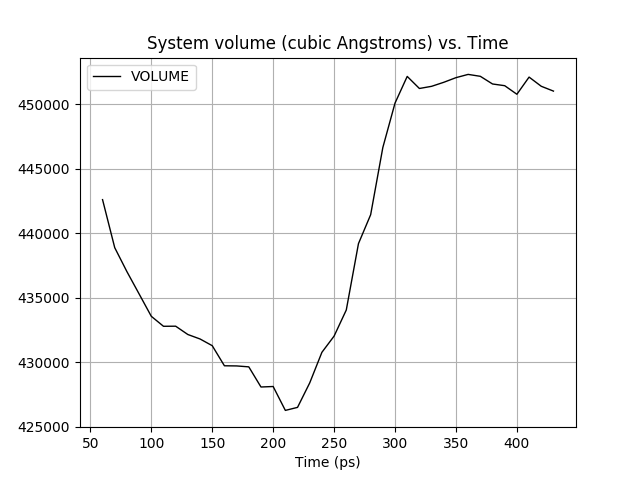
(image/png attachment: rel2_volume.png)
(image/png attachment: rel3_volume.png)
(image/png attachment: prod1_volume.png)
