Date: Fri, 1 Jun 2018 11:26:20 -0300
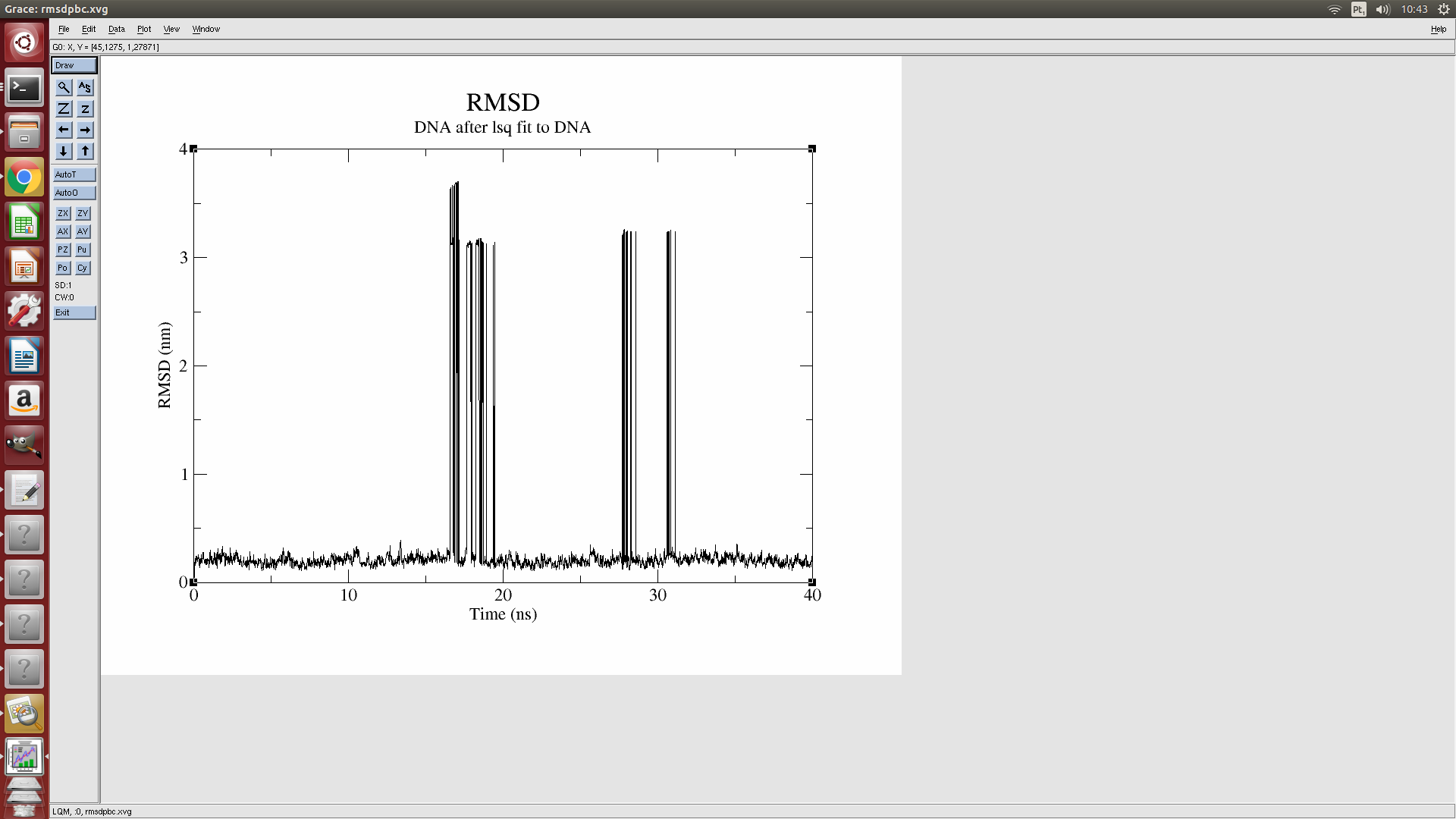
I performed a simulation between a small molecule and B-DNA. The result of
the RMSD is this of the figure. By analyzing the structure at these points,
it appears that some atoms get far away from the DNA, inside the box. Why
does this happen? Does this make my simulation invalid?
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2018-06-01_10:43:30.png)
