Date: Fri, 23 Feb 2018 15:39:06 -0300
Dear from the AMBER list,
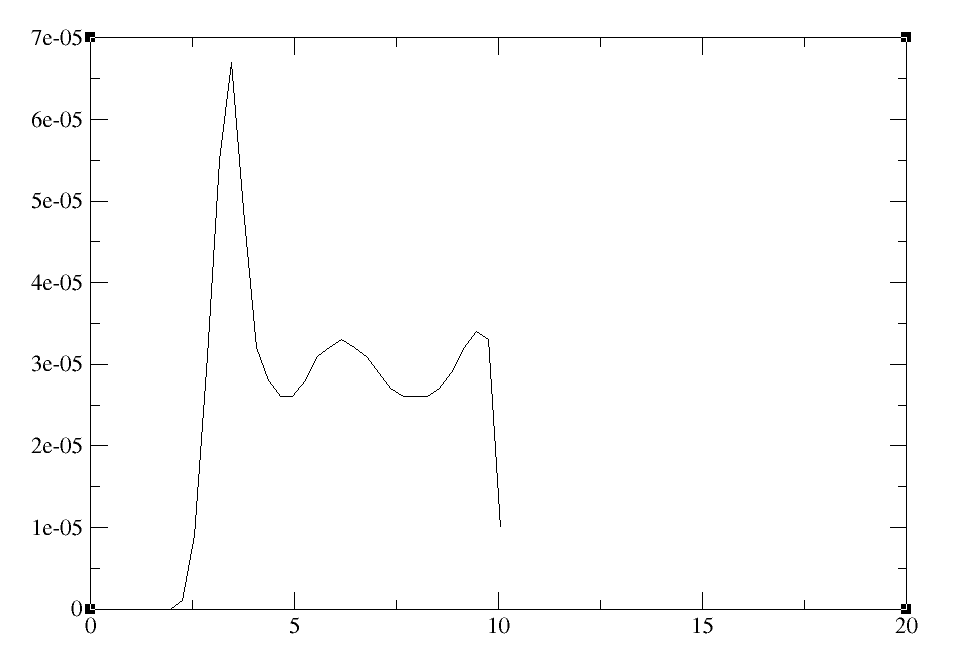
I am getting the RDF from a specific atom of the active site of
a protein.
my simulation in the field of production is 800 ns
I'm using the following script with cpptraj and getting the RDF profile
below:
___________________________________________________________________________
trajin estagio_14.mdcrd
autoimage
radial radial.dat 0.3 10.0 :332.H1 :WAT.O&!(:1-332.O) density 0.022 intrdf
radial.dat
___________________________________________________________________________
[image: Imagem intercalada 1]
My question is: how do I define the number of water molecules in
each layer of solvation obtained on average, for example?
Best regards,
Marcelo
Marcelo Andrade Chagas, MSc
(PhD student)
Laboratório de Química Computacional e Modelagem Molecular - LQC-MM
* http://lqcmm.qui.ufmg.br/
Departamento de Química da Universidade Federal de Minas Gerais - UFMG
Tel:(31)3409-5776
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
