Date: Thu, 28 Sep 2017 16:31:40 +0530
thank you for your kind response.I am uploading two pictures.One is
initial structure and the other is at 32 ns. From the two structure it is
clear that alpha helices and 3-10 helices are already started to be
unfolded. And sometimes beta sheet are appearing. I further run for 60ns.
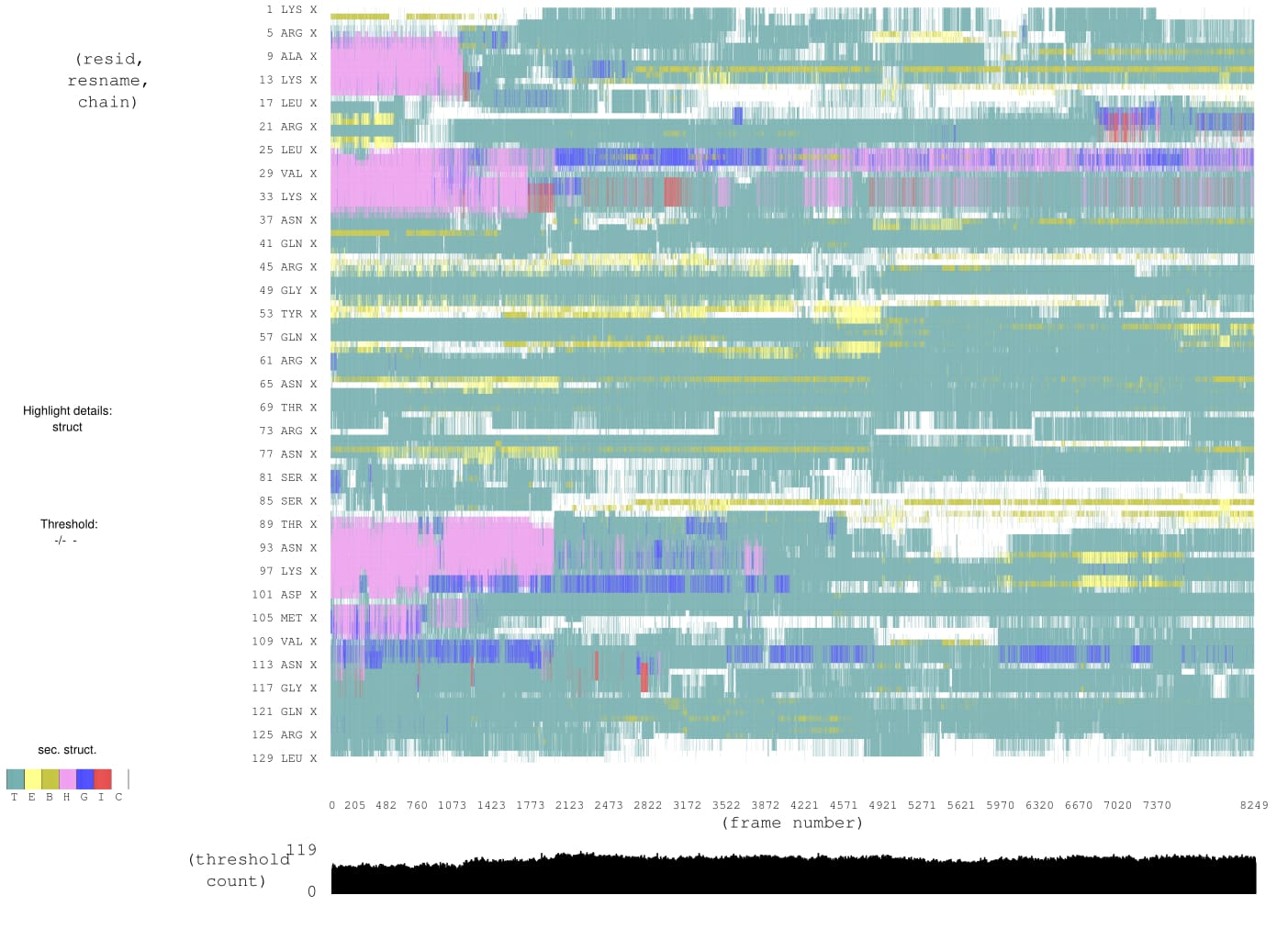
At 60ns I calculated the percentage of helicity. I found percentage of
helicity is too low at 60ns.That means it is unfolding.But Rg value is
still around 13.8A. Now from your suggestion I can assume that though
unfolding is occurring but the local native contacts(distance between the
alpha-carbons)are not being totally disrupted and the protein structure is
not being stretched.As the distance is not increasing so the compactness
will not change. That means, it will be also true for Solvent Accessible
Surface Area(SASA) of that protein.
Thanking You
Krishna
> Hi,
>
> On Tue, Sep 26, 2017 at 12:07 PM, <krishna.2015.iitg.ernet.in> wrote:
>> not happen at all. When a protein unfolds totally then its Rg
>> value should go beyond 17-18A which is reported in many
>
> Just because you have a high RMSD and changes in secondary structure
> this does not mean you have complete unfolding. High RMSD just means
> you are far away from whatever your reference structure is. The
> protein could just be adopting a different conformation (or set of
> conformations). Your best bet is to actually visualize what is
> happening to your system with your favorite visualization program. I
> often find that observing trajectory data can reveal details that are
> easily lost in comparatively simple plots.
>
> -Dan
>
>> literatures. If you kindly check my inputs whether I am doing
>> right or wrong as I am doing these by CPPTRAJ program then it will be
>> great pleasure to me.
>> I have attached the input file as well as the graphs in attachment.
>>
>> I will be highly obliged if you kindly help me to figure out the
>> problem.
>>
>> Thanking you.
>>
>> Krishna
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
>
>
> --
> -------------------------
> Daniel R. Roe
> Laboratory of Computational Biology
> National Institutes of Health, NHLBI
> 5635 Fishers Ln, Rm T900
> Rockville MD, 20852
> https://www.lobos.nih.gov/lcb
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
Krishna Gopal Chattaraj
Research Scholar
Chemistry Department
IIT Guwahati
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/postscript attachment: VMD_Timeline_Window_initial.eps
(image/jpeg attachment: VMD_Timeline_Window_8250-1.jpg)
