Date: Sat, 23 Sep 2017 22:05:26 +0500
Dear Users
I use this command to prepare the .com input file for g09 antechamber -i
name.pdb -fi pdb -o bay.com -fo gcrt
for optimization this basis set and theory use in the route section
#HF/6-31G* SCF=XQC Test Pop=MK iop(6/33=2) iop(6/42=6) opt
and simulation terminate normally. check point file and log file created.
Then use these g09 output file for resp input file by using this command
antechamber -i bay.log -fi gout -o bay.mol2 -fo mol2 -c resp
this give following error
antechamber can only handle one unit. If the input is a single unit
then the connectivity is wrong and the geometry may be bad.
Please convert your molecule to a mol2 file via:
antechamber -j 5 -at sybyl -dr no
And then check your molecule with a visualization program;
manually add missing bonds or delete unwanted bonds as appropriate.
Then I opened the log file structure to visualize it. The structure break
not look in its original given form. I also attached the images before opt
and after opt. Due to this break of structure two unit show that cause
antechamber error for RESP.
Kindly guide
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
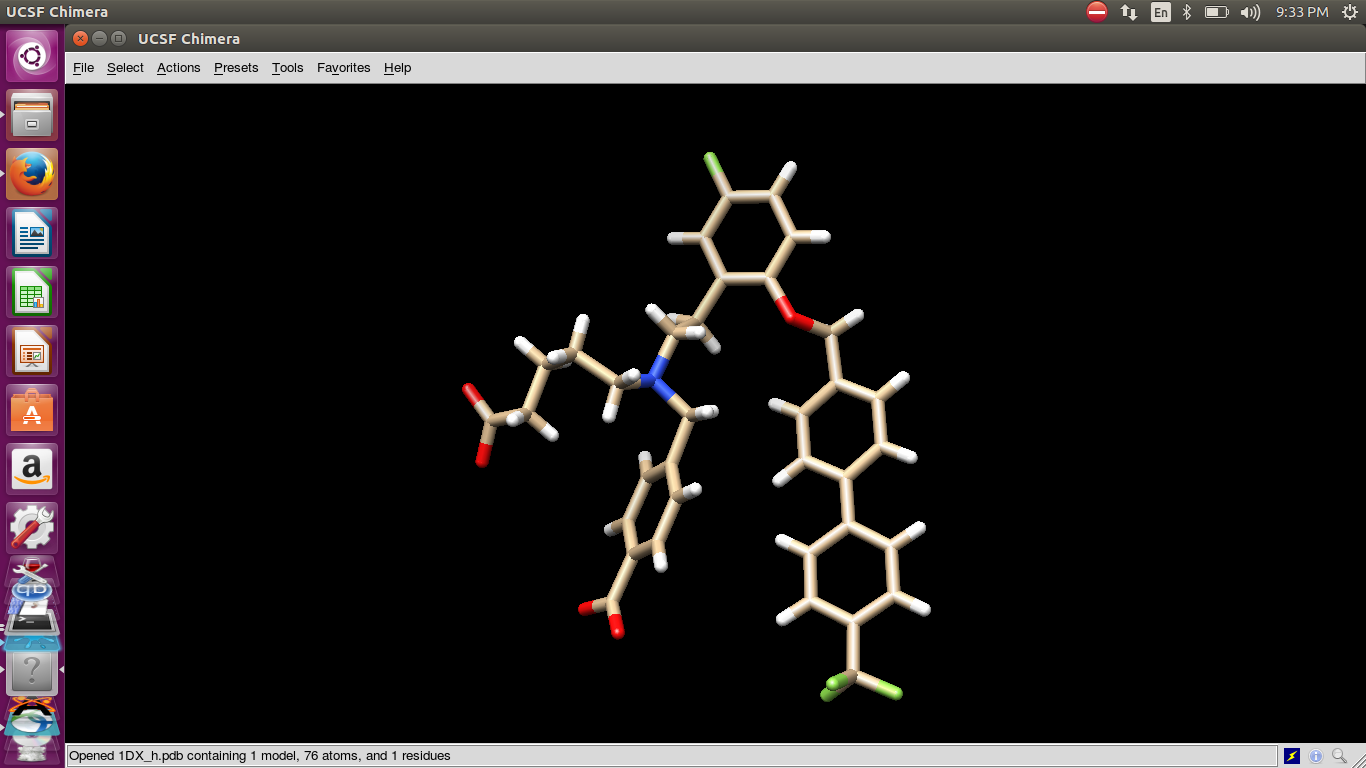
(image/png attachment: bay_before_opt.png)
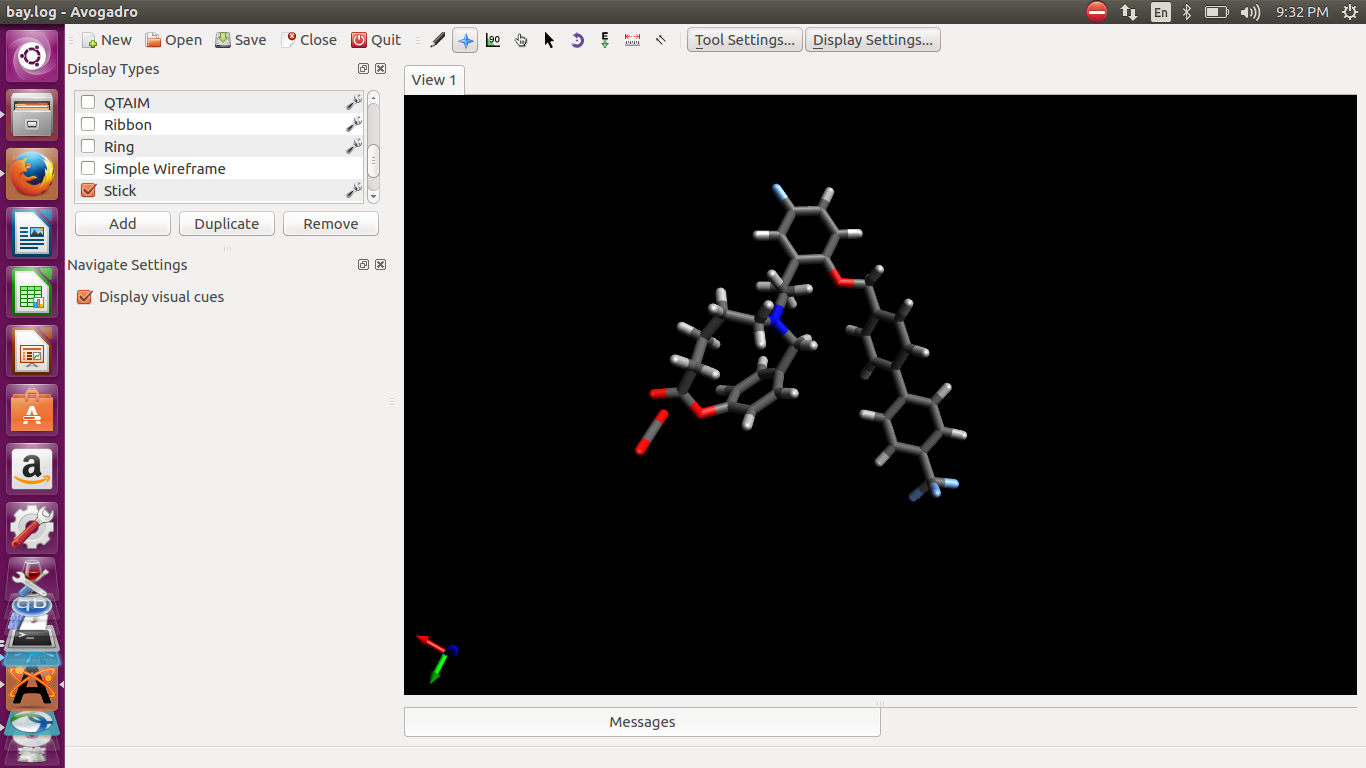
(image/png attachment: bay_after_opt.png)
